
Abstract
Tumor-homing peptides with tissue-penetrating properties increase the efficacy of targeted cancer therapy by delivering an anticancer agent to the tumor interior. LyP-1 (CGNKRTRGC) and iRGD (CRGDKGPDC) are founding members of this class of peptides. The presence of the cysteines forming the cyclizing disulfide bond complicates conjugation of these peptides with other molecules, such as drugs. Here, we report the synthesis of conjugatable disulfide-bridged peptides and their conjugation to biologically important molecules. We have synthesized the LyP-1, iRGD, and CRGDC (GACRGDCLGA) peptides with a cysteine or maleimidohexanoic acid added externally at N-terminus of the sequences. Subsequent conjugation to payloads yielded stable compounds in which the tumor-homing properties of the peptide and the biological activity of the payload were retained.
Keywords
Introduction
Synaphic (docking-based, ligand-mediated, active) targeting is a promising approach that uses targeting elements, such as homing peptides, for selective delivery of drug payloads to diseased tissues.1––5 These peptides bind to various cell surface receptors, eg, integrins, that are specifically expressed in tumors. These targeting systems largely depend on the homing efficiency of the peptide used in the peptide–payload conjugates. We have previously targeted tumors with a blood clot-binding pentapeptide, CREKA (Cys–Arg–Glu–Lys–Ala), which we coupled to magnetically active iron oxide nanoparticles through a thioether linkage using the N-terminal cysteine.6,7 We have also utilized the CREKA targeting system for atherosclerotic plaques. 8 Members of another class of tumor-homing peptides from our laboratory possess tissue-penetrating properties, and as a consequence, particularly efficiently accumulate in the tumor tissue.9––13 These peptides initially bind to a primary, tumor-specific receptor and are then proteolytically cleaved, acquiring an affinity for a second receptor, neuropilin-1 (NRP-1), which triggers the tissue-penetration process. The primary receptor for LyP-1 is p32, a mitochondrial protein that is frequently upregulated and expressed at the cell surface in tumors, particularly in breast cancers. iRGD, 14 like other RGD peptides, initially binds to αv integrins, which are specifically expressed on tumor endothelial cells (and most tumor cells), and like LyP-1 is then processed to a truncated, NRP-1-binding form.
We wanted to conjugate tumor-penetrating peptides to payloads using strategy similar to the one we used with CREKA. However, these peptides are typically cyclic, and the cyclizing disulfide bond poses problems with regard to the coupling chemistry.
Furthermore, coupling of peptides with functionally important amino acid side chains requires modifications to impart chemoselectivity in order to preserve the biological activity of the peptide.
In the literature,15,16 a number of conjugation protocols have been reported but chemoselective conjugation of disulfide-bridged peptides has not been well studied. The available methods either are not amenable to preparative scale synthesis or have not been described in detail.13,17 Most published methods use carbodiimide reagents to make amide bonds, which are often not chemoselective due to reactive groups, such as amines, acids, and hydroxyls. Other ligation techniques, such as the Staudinger reaction 18 and click chemistry,19,20 use external reagents that could alter the peptide structure or biological activity of the conjugate. Traceless Staudinger 21 and copper-free click reactions 22 use intermediates that are obtained synthetically in low yields and are commercially expensive. Tailor-made syntheses of disulfide-bridged peptides for conjugation, which have been attempted in the literature, either fall short of preparative value or have not been clearly described.1,17,23,24 Feldborg et al 25 synthesized disulfide-bridged peptides with various modifications. Ye et al 26 have reported the synthesis of iRGD with a thiol functionality for optical imaging.
We have also been extensively reporting the application of disulfide-bonded cyclic peptides to direct micelles and paclitaxel-albumin nanoparticles to target tissues, 10 especially breast cancers, and to coat cell culture plates for growing human embryonic stem cells. 27 Herein, we report the methods we have been using to synthesize these modified peptides in our laboratory and their characterization. The activation of the extra cysteine functionality of this modified peptide and its utility in oligonucleotide conjugation is further described here. We show that significant scrambling of disulfide bonds did not occur during the synthesis, the desired products could be isolated in good yields, and the functional properties of the peptides were preserved through the conjugation process.
Methods
General materials
Fmoc-amino acid derivatives with different side-chain protections; the reagents N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate, (HBTU), 1-hydroxybenzotriazole (HOBt), 2,4,6-collidine, piperazine, and trifluoroacetic acid (TFA); and solvents N-methyl-pyrrolidinone (NMP), N,N′-dimethylformamide (DMF), dichloromethane (DCM), and acetonitrile were purchased from commercial sources. Other starting materials, namely, 5(6)-carboxyfluorescein (FAM), 5(6)-carboxytetramethylrhodamine (Rd), and linkers N-(α-maleimidoacetoxy) succinimide ester and maleimide-5KPEG NHS were purchased from various sources. HeLa pLuc 705 cells, kindly provided by R. Kole and B. Leblue, were grown in Dulbecco's modified Eagle's medium with GlutaMAX supplemented with 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 mg/mL streptomycin, and 200 μg/mL hygromycin (Invitrogen) at 37°C, 5% CO2 in an incubator. All animal experiments were performed according to procedures approved by the Animal Research Committee at the University of California, Santa Barbara, Santa Barbara, and the Sanford, Burnham, Prebys Medical Discovery Institute, San Diego.
Peptide synthesis
Peptides were synthesized on solid phase using the Fmoc//Bu strategy on a microwave-assisted automated peptide synthesizer (Liberty, CEM Corporation). The amino acids were coupled using HBTU-collidine reagent system in DMF. Fmoc deprotection was performed with 5% piperazine in NMP containing with two microwave energy cycles for 30 and 180 seconds at 75°C under nitrogen (N2). The coupling of amino acids except Arg and Cys was done for five minutes with 25 W microwave energy at 75°C under N2. Fmoc-Arg(Pbf)-OH was double coupled with 0 W microwave energy for 25 minutes and with 20 W microwave energy for five minutes at 75°C. All the cysteine residues were coupled for a total of six minutes at 50°C with 0 and 20 W microwave energy for two and four minutes, respectively. The modified peptides were synthesized in different methods as described below. The protected peptide resins were then cleaved with TFA:triisopropylsilane (TIS):water (95%:2.5%:2.5%) to yield the crude peptides, which were analyzed by reverse phase high-performance liquid chromatography (HPLC) using Gilson's Analytical HPLC (Phenomenex Jupiter Proteo, 250 mm × 4.60 mm column) and purified on Gilson's Semipreparative HPLC (Phenomenex, Jupiter Proteo, 250 mm × 15 mm column). The solvents used were A, 0.1% TFA in water, and B, 0.1% TFA in 60% acetonitrile in water. The peptides were characterized by electron spray ionization mass spectroscopy (ESIMS) on a Waters Micromass QTOF2 tandem mass spectrometer. The data on different peptides are compiled in Table 1.
Characteristics of peptides synthesized using different methods.

Synthesis method A
The peptides were assembled on rink amide MBHA resin with Fmoc-Cys-(S-tBu)-OH on the C-terminus and Fmoc-Cys(Mmt)-OH on the N-terminus. Furthermore, the third cysteine was introduced in the sequence with Fmoc-Cys(Trt)-OH on the N-terminus spaced from the sequence with 6-aminohexanoic acid. All other residues had standard Fmoc-synthesis compatible protecting groups. FAM was coupled with 25 W microwave energy for five minutes at 75°C under N2, and Rd was coupled manually using DIC–HOBt. After the completion of synthesis, the thiobutyl protection on the C-terminus cysteine was removed using 50 equiv of tri(n-butyl)phosphine (TBP) in 4 mL of NMP:water (9:1). The free thiol, thus liberated, was activated using 2,2′-Dithiobis (5-nitropyridine) (DTNP) in DCM for one hour. The peptide was cyclized by treating the peptide resin with 1% TFA and 5% TIS in DCM two times for 10 minutes each, and the resin was dried. Upon cleavage with 95% TFA, 2.5% TIS, and 2.5% water, the modified peptides were obtained in 20% yields (based on the initial resin loading) after purification. The peptides were purified by HPLC using a solvent system of A, 0.1% TEA in water, and B, 0.1% TEA in a mixture of 60% acetonitrile and 40% water at a flow rate of 10 mL/minute on a Alltima C18 column (250 mm × 22 mm, 10 μm particle size, and 100 A pore size). The peptides were analyzed by HPLC using a solvent system of A, 0.1% TEA in water, and B, 0.1% TEA in a mixture of 60% acetonitrile and 40% water at a flow rate of 1 mL/minute on a Alltima C18 column (250 mm × 4.60 mm, 10 μm particle size, and 100 å pore size). The purified peptide products were characterized by ESIMS-time of flight (TOF) and MSMS-TOF.
Synthesis method B
The peptide was assembled on rink amide MBHA resin with Fmoc-Cys(S-tBu)-OH on the C- and N-termini. The third cysteine was introduced in the sequence with Fmoc-Cys(Trt)-OH on the N-terminus spaced from the sequence by 6-aminohexanoic acid. All other residues had standard Fmoc/tBu-synthesis compatible protecting groups. After the completion of synthesis, the thiobutyl protection on the cysteines was removed using 50 equiv of TBP in 4 mL of NMP:water (9:1). The free cysteines liberated were converted to cystine on the resin using 1 equiv of CBr4 and 2 equiv of Et3N in NMP at room temperature in two hours. Upon cleavage with 95% TEA, as described in method A yielded the conjugatable peptide in 20–25% yields based on the initial resin loading.
Synthesis method C
The peptide was assembled on rink amide MBHA resin with Fmoc-Cys(Acm)-OH on the C- and N-termini. The third cysteine or maleimide was introduced in the sequence with Fmoc-Cys(Trt)-OH on the N-terminus spaced from the sequence by 6-aminohexanoic acid or with €-maleimidocaproic acid. All other residues had standard Fmoc/tBu-synthesis compatible protecting groups. After the synthesis, the peptides were cyclized using thallium(III) trifluoroacetate (Tl(tfa)3) in DMF with 5% anisole scavenger. Upon cleavage with 95% TEA, as described in method A, yielded the conjugatable peptide in 15% yields based on the initial resin loading.
Synthesis of peptides with nitro-pyridine sulfenyl (Npys)-activated cysteine
The peptides with free N-terminal cysteine and 5 equiv of DTNP were dissolved in TFA:water (4:1) and stirred under N2 atmosphere for 15 minutes. The reaction mixture was concentrated and dissolved in water and filtered to remove the insoluble solids. The filtrate was lyophilized and purified by HPLC using a gradient of 40% B–80% B over 45 minutes to yield the final product. The peptide characteristics are listed in Table 1.
Preparation of nanoworms (NWs)
NWs
7
coated with peptides
In vivo NW injections
To analyze NW biodistribution, mice bearing orthotopic 22Rvl tumors were injected with the peptides
Preparation of FAM-X-C(iRGD)REKA conjugate
€-maleimidocaproyl-iRGD peptide
In vivo FAM-X-C(iRGD)REKA conjugate injection
Mice bearing MCF10Ca1A human breast cancer xenograft tumors were intravenously injected with FAM-X-CREKA, FAM-X-iRGD, or FAM-X-C(iRGD)REKA conjugates, and tumor sections were processed as described earlier.
Conjugation of peptide 9 to antisense (as)-RNA
FAM-Cys(oligonucleotide)-X-LyP-1 conjugate (10)
Peptide
Splice correction assay
Cells were plated overnight on a 96-well plate at a concentration of 10000 cells/well. Small quantities of lipofectamine 2002 were complexed with unconjugated nucleotide and peptide-conjugated nucleotide at various concentrations and incubated on the cells in serum-free media. Expression of luciferase was assayed using Luciferase Assay Kit (Promega).
Immunocytochemistry
Splice-corrected cells were fixed using 4% paraformaldehyde and blocked using 1% FBS. The cells were incubated with antiluciferase antibody (1:1000; Promega) for one hour at room temperature. Secondary antibody 647 donkey antigoat (1:1000; Molecular Probes) was used to detect the primary antibody. Each experiment included cells stained with secondary antibodies alone as a negative control. Nuclei were counterstained with DAPI (5 μg/mL; Molecular Probes). The cells were visualized under an inverted fluorescent microscope (Leica DM IRB; Leica Microsystems).
Results and Discussion
Peptide synthesis
Synthesis of the homing peptides was based on a strategy to obtain the final peptide with a free cysteine and the desired disulfide bridge upon cleavage. This was done by selecting cysteine residues with protections that are sensitive to very dilute acid or to reducing agents. Initially, peptides were designed to have an extra N-terminal cysteine separated by a 6-aminohexanoic acid spacer and a fluorophore for imaging that was attached to the extra cysteine. The peptides, LyP-1,
28
iRGD,
14
and a nontumor-penetrating peptide CRGDC (GACRGDCLGA)
29
, were assembled on a microwave-assisted automated peptide synthesizer following Fmoc/tBu strategy on a rink amide MBHA resin. The peptides were labeled with 5(6)-carboxyfluorescein (FAM) or 5(6)-carboxytetramethylrhodamine (Rd). Initially, for the synthesis of iRGD peptides

Directed disulfide bond formation in iRGD peptide with a third cysteine. (a) t-Butylphosphine in NMP:water (9:1), (b) 2,2-dithiobis(5-nitropyridine) (DTNP) in DCM, (c) 1% TFA in DCM, and (d) 95:2.5:2.5.
After the synthesis, peptide resin was treated with 50 equiv of TBP in NMP:water (9:1) twice for one hour each as per Beekman et al protocol. 30 Gongora-Benitez et al 31 observed that proximity of S-Trt groups influenced the S-tBu deprotection resulting in incomplete or no deprotection with DMF as the solvent. We have observed a complete removal of S-tBu groups in NMP–water mixture though the cysteine was on the C-terminus. The resin-bound partially deprotected peptide resin was reacted with 10 equiv of DTNP in DCM to activate the free thiol and cyclized using 1% TFA in DCM. 32 Cleavage using a standard 95% TFA cocktail released the peptide containing both the intended disulfide bridge and a free cysteine as intended in 15–20% yields after purification.
Looking to improve the synthesis, we adapted bisprotection strategy, wherein the two cysteines in the original sequence were protected with S-tBu and the external cysteine was protected with Trt group. After the synthesis, the S-tBu groups were removed using 50 equiv of TBP in NMP:water (9:1) twice for one hour each as done earlier for one S-tBu. Test cleavage and monitoring the deprotection with ESIMS indicated the removal of both the S-tBu groups. Subsequently, the partially deprotected peptide was cyclized using a modification of a literature method.
33
The resin-bound partially deprotected peptide was treated with 1 equiv of CBr4 and 2 equiv of Et3N. The standard cleavage yielded the desired free cysteine peptide

Disulfide bond formation in LyP-1 peptide using CBr4. (a) t-Butylphosphine in NMP:water (9:1), (b) CBr4 (1 equiv), and (c) TEA:TIS:water (95:2.5:2.5).
The structures of the peptides synthesized using the above methods were confirmed by mass spectrometry as shown in Table 1. ESIMSMS spectra of cysteine-modified peptides
Seeking to improve the method further, we chose S-acetamidomethyl (S-Acm) protection for the cyclizing cysteines, and a Trt group was used for the third cysteine. The S-Acm-protecting groups were removed, and the cysteines were cyclized in situ using Tl(tfa)3. For this protection, Tl(tfa)3 was the reagent of choice for deprotection and in situ cyclization. The cyclization was completed at 0°C in seven to nine hours depending on the sequence. We have not found the scrambled disulfide products that may be expected from the cleavage of Cys(Trt) in detectable levels. This selectivity in reactivity of Trt group toward Tl(tfa)3 was found to be in agreement with Albericio et al.
34
The three methods all aimed at obtaining a peptide that has a disulfide bridge and an extra cysteine gave similar yields and purities. So any one of the methods can be chosen for synthesizing these modifications. Furthermore, we have replaced the cysteine and fluorescein at the N-terminus with EMCA to obtain the maleimide-functionalized iRGD (

Disulfide bond formation in LyP-1 peptide with a third cysteine. (a) Ti(OTfa)3 DMF:anisole (9:1) and RT (nine hours) and (b) TFA:TIS:water (95:2.5:2.5).
In order to increase the rate and selectivity of the conjugation, we chose to activate the extra cysteine in the peptide with a Npys group to facilitate an asymmetric disulfide bond formation with a thiol group-bearing cargo. This strategy aids a heterodimeric disulfide formation at a faster rate and at low pH, which minimizes cysteine scrambling. We modified the method reported by Rabanal et al
35
for heterodimerization. We treated peptides

Synthesis of Npys-activated disulfide-bridged peptides for intracellular payload delivery.
The peptide products were >95% pure by HPLC and showed the expected characteristics in ESIMS. The peptides labeled with 5(6)-FAM showed four well-resolved peaks corresponding to four geometrical isomers (Supplementary Files 1–4). This is due to the positioning of the Npys substitutions relative to FAM on the cysteine α-amino group across the plane of symmetry along the disulfide bond between the cysteine and the Npys groups. This was confirmed by synthesizing the Npys-activated peptide iRGD without the FAM label, which appeared as a single peak in the HPLC.
Peptide conjugation through Michael addition
To examine peptide stability, peptide
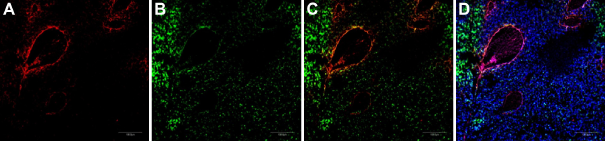
Combining CRGDC NWs with LyP-1 NWs enhances homing to different areas of tumor. A mixture of iron oxide NWs coated with rhodamine-labeled CRGDC or FAM-labeled LyP-1 was intravenously injected (2.5 mg iron/kg of each NW preparation) into nude mice bearing orthotropic 22Rv1 tumors. The mice had been preinjected with Ni liposomes to reduce uptake by the reticuloendothelial system. 7 The mice were perfused through the heart five hours later, tumors were harvested and sectioned, and the sections were examined by confocal microscopy. Nuclei were stained with DAPI (blue). Scale bars, 200 μm. (A) CRGDC (peptide 5) NWs (red), (B) LyP-1 (peptide 3) NWs (green), (C) panels (A) and (B) superimposed, and (D) DAPI (blue) costaining with C.
To test the effect of the maleimide functionalization, maleimide-iRGD (peptide

Homing of peptide-coated nanoworms to breast cancer xenografts. Mice bearing MCF10Ca1A human breast cancer xenograft tumors were intravenously injected (A) FAM-X-CREKA, (B) FAM-X-iRGD, or (C) FAM-X-C(iRGD)REKA conjugates, and tumor sections were processed as in Figure 5. NWs are green; blood vessels visualized with anti-CD31 are red. Arrows point to vascular structures that show colocalization of nanoparticle and CD31 signals. FAM-X-CREKA is associated with blood vessels, but because it binds to clotted plasma proteins within tumor vessels, 6 there is little colocalization (yellow color) with the endothelial cells marked with CG31. iRGD causes extravasation into tumor parenchyma of the other two peptides, resulting in even less blood vessel localization than seen with FAM-X-CREKA. Nuclei were stained with DAPI (blue). Representative results from three experiments are shown.
Conjugation through a disulfide link
A disulfide linkage is well suited for intracellular delivery, as it is cleaved by cytoplasmic glutathione, releasing the active payload. The Npys-activated product

Intracellular oligonucleotide delivery to HeLa cells. (A) Gel electrophoresis of oligonucleotides–-channels: (i) as-RNA, (ii) LyP-1-as-RNA conjugate (Compound 10), and (iii) as-RNA and LyP-1 peptide mixture 10. (B) Confocal micrographs showing expression of luciferase upon delivery of Compound 10. (C) as-RNA without LyP-1 conjugated to it causes no luciferase expression. (D) Dose–response curve for luciferase expression upon Compound 10 addition.
Conclusion
In summary, we have successfully synthesized and demonstrated the structural and functional integrity of disulfide-bridged peptides with a free thiol or maleimide functionalities for conjugation through thioalkyl and disulfide linkages. We have demonstrated their conjugation to different cargoes, and that the nanoparticle, and oligonucleotide, conjugates have the desired activities and efficacies. These results establish chemoselective conjugation of peptides having sensitive disulfide bridges to a variety of payloads. Thus, this is a powerful tool for disulfide-bridged peptide-mediated imaging, targeted drug delivery, and other applications.
Footnotes
Author Contributions
Conceived and designed the experiments: VRK and ER. Contributed data and data analysis: VRK, SS, PK, LA, JP, and ER. Wrote the first draft of the article: VRK. Contributed to the writing of the article: VRK, SS, PK, LA, and ER. Agree with the article results and conclusions: VRK, SS, PK, LA, JP, and ER. Jointly developed the structure and arguments for the article: VRK, SS, PK, LA, JP, and ER. Made critical revisions and approved the final version: VRK and ER. All authors reviewed and approved the final article.