
Abstract
Pathogenic mutations in BRCA1 or BRCA2 are only detected in 25% of families with a strong history of breast cancer, though hereditary factors are expected to be involved in the remaining families with no recognized mutation. Molecular characterization is expected to provide new insight into the tumor biology to guide the search of new high-risk alleles and provide better classification of the growing number of BRCA1/2 variants of unknown significance (VUS). In this review, we provide an overview of hereditary breast cancer, its genetic background, and clinical implications, before focusing on the pathologically and molecular features associated with the disease. Recent transcriptome and genome profiling studies of tumor series from BRCA1/2 mutation carriers as well as familial npn-BRCA1/2 will be discussed. Special attention is paid to its association with molecular breast cancer subtypes as well as the latest advances in predicting BRCA1/2 involvement (BRCAness) using molecular signatures, for improved diagnostics and selection of patients sensitive to targeted therapeutics.
Introduction
Breast cancer is the most frequent malignant disease and the leading cause of cancer death among women in both economically developed and developing countries. Globally, 1.4 million new breast cancer cases are diagnosed each year, of whom approximately one-third die of the disease. 1 The incidence rates are highest in the Western world, where the lifetime risk of developing breast cancer is estimated to be one in nine. Owing to increased awareness, early detection, and better treatment options available, breast cancer mortality rates have declined in recent years. 2
In the middle of the 19th century, the first reports emerged, describing familial aggregation of breast cancers. 3 Today, positive family history is one of the most important risk factors for developing breast cancer. It is currently estimated that approximately 5–10% of all breast cancers have a hereditary background. These families show an apparently dominant inheritance pattern and are often characterized by an early age of onset, overrepresentation of ovarian cancers, bilateral breast cancers, and male breast cancers. 4
BRCA1- and BRCA2-Associated Breast Cancer
Early reports suggested that germline mutations in the genes BRCA1 and BRCA2 were responsible for the majority of hereditary breast cancers, although more recent studies have demonstrated that mutations in the two genes only account for 25–28% of the family risk.5,6 However, it is expected that additional BRCA1/2 mutations remain undetected by the screening methods used today. Women carrying a BRCA1 or BRCA2 germline mutation also have increased risk of developing ovarian cancer and fallopian tube cancer. In addition, BRCA2 mutation carriers also have increased risk of other cancer types such as male breast cancer, prostate cancer, pancreas cancer, gastrointestinal cancers (gall bladder, bile duct, and stomach), and melanoma.7–9 In a large study by the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA), the median age of diagnosis was found be to be 40 years among BRCA1 and 43 years among BRCA2 mutation carriers. 10
Even though germline mutations in BRCA1 and BRCA2 confer high risk of breast and ovarian cancers, the penetrance of these genes is incomplete. The risk in BRCA1 and BRCA2 mutation carriers of developing breast cancer by the age of 70 is 45–87%. For ovarian cancer, the risk is 45–60% among BRCA1 mutation carriers and 11–35% among BRCA2 mutation carriers.11–14 However, the penetrance depends on several different factors, including the type of mutation and exogenous factors. Lifestyle factors such as physical exercise and lack of obesity in adolescence have been associated with significant delay in breast cancer onset. 11 It has been shown that common breast cancer susceptibility alleles may act multiplicatively on the breast cancer-risk in BRCA1 and BRCA2 mutation carriers, which might explain why the risk seems to be highest in women from families with multiple breast cancer cases. 15
Both BRCA1 and BRCA2 have considerable complex genomic structures, and the coding regions show no homology to previously described genes or to each other. The BRCA1 gene is composed of 24 exons encoding a very large protein of 1,863 amino acids, while BRCA2 consists of 27 exons encoding an even larger protein of 3,418 amino acids. In both genes, the first exon (exon 1) is non-coding and exon 11 is remarkably large.16,17
BRCA1 and BRCA2 function as tumor suppressor genes and are important in maintenance of genomic stability through their role in DNA damage signaling and DNA repair. Both BRCA1 and BRCA2 are implicated in mediating repair of double strand breaks by homologous recombination (HR) by interactions with RAD51. Upon DNA damage, BRCA1 will associate with RAD51 and localize to the damaged region by which BRCA1 becomes phosphorylated. BRCA2 functions downstream of BRCA1 by complex-formation with RAD51. The primary function of BRCA2 is to facilitate HR. 18 Cells deficient for BRCA1 or BRCA2 are unable to repair double strand breaks by the error-free HR, resulting in repair by the error-prone non-homologous end-joining (NHEJ) pathway introducing chromosomal instability.19,20 During S-phase, the expression levels of BRCA1 and BRCA2 increase, indicating a function in maintaining genomic stability during the DNA replication process. 21 Besides its role in HR, BRCA1 appears to have additional functions in DNA repair. BRCA1 is also part of the BRCA1-associated genome-surveillance complex (BASC), which includes ATM, RAD50, MRE11, and NBS1 and the mismatch repair proteins MLH1, PMS2, MSH2, and MSH6. 22 BRCA1 has also been demonstrated to be involved in transcription-coupled excision repair, chromatin remodeling, and together with BARD1 in the ubiquitination process, by which proteins are tagged for degradation by the proteasome.18,23
In all, 1,790 distinct mutations, polymorphisms, and variants in the BRCA1 gene and 2,000 in BRCA2 have been reported to the Breast Cancer Information Core (BIC) database, respectively (July 2014). 24 Approximately 53–55% of these are private mutations, are only detected in single families. Mutations are distributed across the entire coding sequences. The most common types of pathogenic mutations are small deletions or insertions or nonsense mutations resulting in protein truncation leading to non-functional protein. Mutations affecting splice-sites as well as large genomic rearrangements are also observed in both genes.8,25 Missense mutations, silent mutations, and polymorphisms are also frequently identified; however, the clinical interpretation of their pathogenic potential is often difficult. Also, variants such as small in-frame insertions and deletions and possible splice-site alterations are problematic for precise cancer-risk estimation. Almost 1,800 distinct sequence variants found in BRCA1 and BRCA2 are classified as having unknown clinical significance (unclassified variants, UVs). To assess the clinical significance of individually rare sequence variants is challenging, as existing methods require a high number of occurrences of the specific variant. In 2009, the ENIGMA (Evidence-based Network for the Interpretation of Germline Mutant Alleles) consortium consortium was established with the purpose of evaluating the clinical significance of rare sequence variants by pooling genetic and associated clinical and histopathological information from a world-wide network of laboratories to gather sufficient data and resources to facilitate the classification of UVs. 26
A germline mutation in BRCA1 or BRCA2 only represents the first hit in the classical Knudson's two-hit hypothesis, whereas the second inactivating somatic mutation often involves deletion of the wild-type allele, termed loss of heterozygosity (LOH). LOH has been reported to be present in the majority (≫80%) of tumors arising from mutation carriers.27,28 In contrast, small somatic mutations involving a single or few bases are very rare. 29 Another somatic inactivation mechanism, epigenetic silencing by promoter methylation, has been reported of BRCA1 in 9–13% of sporadic breast tumors, an up to 42% in non-BRCA1/2 hereditary breast tumors leading to reduced BRCA1 expression.30–34 In contrast, BRCA1 promoter methylations are rare in tumors from BRCA1 and BRCA2 mutation carriers, 35 and BRCA2 promoter methylation in general is seldom observed in both sporadic and hereditary breast cancers. 36
Familial non-BRCA1/2 Breast Cancer
Several rare gene variants have been described to confer an increased risk of breast cancer, involving high-penetrance genes such as TP53, CDH1, PTEN, STK11, RAD51C, and RAD51D and the low/moderate-penetrance genes such as ATM, CHEK2, BRIP1, and PALB2, among others (reviewed by Vargas et al). 37 In general, most of these genes are involved in the maintenance of genomic integrity and DNA repair mechanisms, and many are associated with multiple cancer syndromes such as Li–Fraumeni syndrome (TP53), Cowden syndrome (PTEN), and Peutz–Jeghers syndrome (STK11/LKB1).38–40 Furthermore, a number of common low-penetrance breast cancer alleles have recently been identified by genome-wide association studies (GWAS), including 10q26, 16q12, 2q35, 8q24, 5p12, 11p15, 5q11, and 2q33.41–43
Low- and moderate-penetrant genes/loci can only explain a minor fraction of the remaining non-BRCA1/2 families that show high incidence of breast cancer. Despite intensive research, genetic linkage analysis, GWAS, and most recently, next-generation sequencing (NGS) exome studies have failed to identify other common high-penetrance breast cancer susceptibility genes, such as BRCA1 and BRCA2, and more than 70% of the genetic predisposition to breast cancer remains unexplained. No single high-penetrance gene is likely to account for a larger fraction of the remaining familial aggregation.44–49 Instead, the remaining predisposition is expected to be a mixture of rare high-risk variants and polygenic mechanisms involving more common and/or rare low-penetrance alleles or rare moderate-penetrance genes, acting in concert to confer a high breast cancer-risk. 38 However, very recently germline mutations in RAD51C have been linked to high cancer-risk in a small number of hereditary breast and ovarian cancer (HBOC) families, supporting the hypothesis that some proportion of the remaining predisposition may be caused by rare high-risk alleles. 50
Eventually, exogenous factors such as oral contraceptive, hormone replacement therapy, alcohol consumption, overweight, and physical inactivity are all known breast cancer-risk factors. 51 An unknown fraction of these families with apparently strong family history could be attributable to such environmental risk factors or could, as breast cancer is a common disease, be random aggregation of sporadic breast cancer cases.
Pathological Characteristics of Hereditary Breast Cancer
The majority of invasive breast cancers arising in BRCA1 and BRCA2 carriers are invasive ductal carcinomas (IDC) (≫80%). A higher frequency of BRCA1 tumors are classified as medullary carcinomas compared to sporadic tumors (9% versus 2%).10,52 Medullary carcinomas are poorly differentiated, high-grade carcinomas with diffuse lymphocytic infiltrate but with a remarkably favorable prognosis, probably because of low incidence of lymph node metastasis. 53 Notably, 11% of medullary carcinomas carry BRCA1 germline mutations. 54 By contrast, excess of invasive lobular and tubular carcinomas has been reported for BRCA2 relative to BRCA1 tumors.10,55 BRCA1 tumors are more frequently high-grade compared to sporadic tumors. They have a higher number of mitosis, and show a high frequency of necrotic areas and a higher proportion of continuous pushing margins and lymphocytic infiltration. All these features point toward a more aggressive tumor type.56,57 Most BRCA2 tumors are grade 2/3 with high mitotic rates. Continuous pushing margins are also characteristic of BRCA2 tumors.
Breast tumors express a number of immunohistochemical (IHC) markers providing both prognostic and predictive information. The estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor (EGFR) 2 (HER2) are among the most important IHC markers. Among sporadic tumors, 70% are ER-positive and 50% are PR-positive, and HER2-overexpression is observed in approximately 15% of the cases. ER-positive tumors respond better to endocrine anti-estrogen treatment, whereas tumors overexpressing HER2 respond well to targeted therapy such as trastuzumab (Herceptin). Approximately 20% of all breast cancer cases are negative for ER, PR, and HER2, known as “triple-negative” (TN) cancers. The prognosis of TN tumors is very poor, not only because these tumors seem to be more aggressive than other breast cancers but also because endocrine and anti-HER2 therapies are ineffective, leaving chemotherapy as the only treatment option available.58,59
A recent study examining pathology data from 4,325 BRCA1 and 2,568 BRCA2 mutation carriers reported that 78% of tumors arising in BRCA1 carriers were ER-negative, while only 23% of tumors arising in BRCA2 mutation carriers were ER-negative. Furthermore, HER2-overexpression was only observed in approximate 10% of the tumors from mutation carriers. Consequently, 69% of the BRCA1 tumors were TN, which was true for only 16% of the BRCA2 tumors. 10 The relation between BRCA1 mutations and low expression of the hormone receptors is significantly different from sporadic tumors even when adjusting for the younger age of the BRCA1 patients. The majority of BRCA1 tumors exhibit a basal/myoepithelial phenotype by expressing several basal markers including the cytokeratins CK5/CK6, CK14, caveolin, vimentin, laminin, p-cadherin, oesteonectin, and the EGFR.56,60 It has also been reported that BRCA1 tumors stained more often p53-positive compared to sporadic, and this probably reflects the higher frequencies and distinct patterns of somatic TP53 mutations that are found among BRCA1 tumors.61,62 As BRCA1 or BRCA2 inactivation leads to cell cycle arrest because of activation of p53, mutations in the TP53 gene have been suggested as a mechanism to escape cell cycle arrest. 63 Several attempts have been conducted where IHC profiles have been applied in combination with morphological characteristics to identify patients with a high risk of carrying a BRCA1 or BRCA2 mutation for clinical classification of unclassified sequence variants, but with mixed success.60,64–66
In contrast to BRCA1 tumors, BRCA2 tumors seem to be more similar to sporadic tumors with relation to the expression of IHC markers. Most BRCA2 breast tumors show a luminal phenotype by overexpressing ER and PR and the cytokeratins CK8 and CK18. 64
Familial non-BRCA1/2 breast cancers have been shown to comprise a very heterogeneous group of cancers with respect to histopathological characteristics. It has been established that these cancers are often of lower grade compared to sporadic cancers, but with IHC profiles similar to sporadic cancers.33,67 Results from studies on breast cancers from CHEK2 mutation carriers have been inconsistent. Two studies found tumors from CHEK2 carriers to be more frequently ER-positive, while one study reported no difference between carriers and non-carriers.68–70
Clinical Implications of Hereditary Breast Cancer
Genetic counseling and risk assessment
Familial breast cancer cases are today identified by evaluation of a family pedigree showing breast and ovarian cancer cases. Presymptomatic testing for pathogenic mutations in BRCA1 and BRCA2 has become widespread during the last decade and is now used in the counseling of families with a strong history of breast and ovarian cancers and for estimating the cancer-risk of healthy family members.71,72 Mutation carriers are recommended intensive surveillance programs of breast and ovaries and offered prophylactic surgery. Prophylactic mastectomy has been shown to lower the risk of breast cancer among mutation carriers. Furthermore, the women are offered prophylactic bilateral salpingo-oophorectomy (BSO) after child-bearing age to lower their risk of developing both breast and ovarian cancers, even further.73–76 Just as important is the fact that if no mutation is detected in a family member of a known BRCA mutation-carrying family, the individual's risk of cancer is equal to that of the general population. Genetic testing of BRCA1 and BRCA2 is often laborious and complex because of the size of the genes. Though newer methods such as targeted NGS have improved the sensitivity, it is likely that a fraction of the mutations remains undetected. In addition, a recent Polish study has demonstrated that up to half of BRCA1 and BRCA2 mutation carriers lack an obvious family history and will therefore not be identified by current selection criteria. 77 As described above, in ≫70% of families with aggregation of breast and ovarian cancers, pathogenic mutation in BRCA1 or BRCA2 cannot be identified. Consequently, the cancer-risk assessment becomes less accurate because of lack of presymptomatic testing options. In addition, unclassified sequence variants are often detected in the coding or non-coding regions of BRCA1 and BRCA2. The clinical significance of such variants is often uncertain and therefore remains a challenge in counseling and clinical management. Confident classification of these variants as well as identification of more high-risk alleles would provide a more accurate risk assessment and improve genetic counseling dramatically for this group of families.
Novel targeted treatment strategies for hereditary breast cancer
As inactivation of BRCA1 and BRCA2 leads to impaired HR DNA repair, it has been investigated whether mutation carriers would be sensitive to DNA cross-lining agents such as platinum salts, as they introduce double-strand DNA breaks. Very encouraging, high response rates to cisplatin have recently been demonstrated in patients with BRCA1 germline mutations.78,79
A novel potential targeted treatment strategy for breast cancer patients with BRCA1 or BRCA2 germline mutations that recently has emerged is the use of poly(ADP-ribose) polymerase (PARP) inhibitors. PARP1 is involved in base excision repair (BER) mechanisms, and inhibition of PARP1 leads to spontaneous single-strand DNA lesions. During DNA replication, these DNA nicks can degenerate to form double-strand breaks during DNA replication because of collapsed replication forks, which activate HR repair. As described previously, inactivation of BRCA1 or BRCA2 leads to impairment of the HR DNA repair pathway, sensitizing the cancer cells to PARP1 inhibition. Disabling both pathways results in chromosomal instability, cell cycle arrest, and apoptosis. Cell survival assays have showed that cells with BRCA1 or BRCA2 inactivation were highly sensitive to PARP-inhibitors.80–82 Early clinical trials demonstrated significant efficiency of PARP-inhibitors in BRCA-deficient breast and ovarian cancers.83–85 Because of the phenotypic similarities between BRCA1-associated and TN cancers, a phase 2 study has been conducted to test the efficiency of iniparib (in addition to standard chemotherapy) in metastatic TN cancers, with promising results. 86 However, the clinical phase 3 trial failed to show significant improvements. BRCA1 and BRCA2 statuses were not assessed; it is therefore not possible to conclude whether a subgroup of BRCA1- and BRCA2-deficient tumors would have benefited from the treatment. This emphasizes the need for more refined methods of selecting patients who will respond to PARP-inhibitors. 87 If targeted therapies against BRCA1- and BRCA2-deficient tumors such as cisplatin or PARP-inhibitors enter clinical practice, genetic testing becomes increasingly important to identify patients with BRCA-deficient tumors.
Molecular Profiling of Hereditary Breast Cancer
During the last decades, the microarray technology has been used extensively to study breast cancer biology. Numerous studies have used the platform for transcriptome and genomic profiling analyses, and recently, studies of genome-wide microRNA and methylations profiling have emerged.88–93 Microarray-based molecular profiling studies have uncovered the complexity and heterogeneity of breast cancer and established that breast cancer is not a single disease entity but rather a group of distinct disorders.
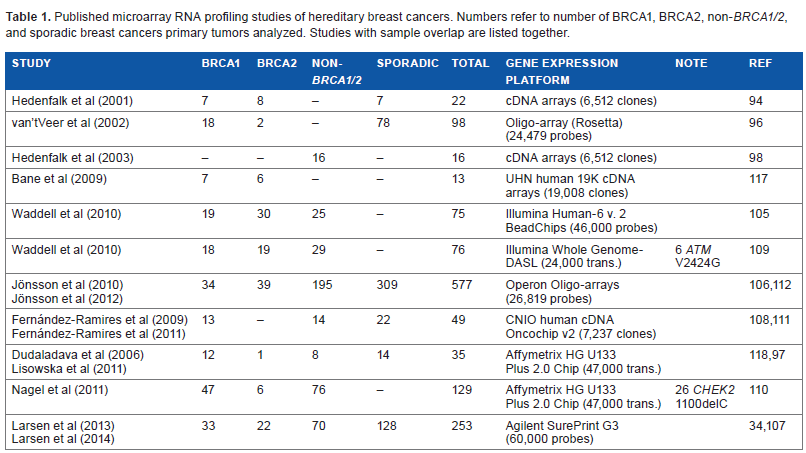
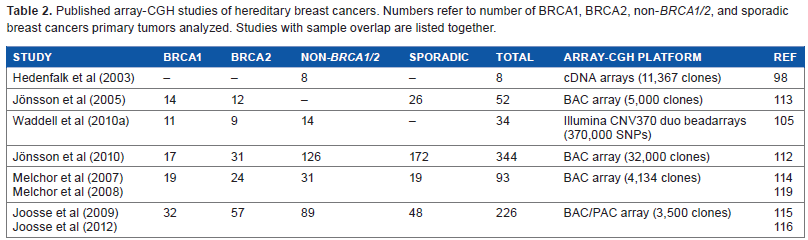
Although an exhaustible number of microarray profiling studies of breast cancers have been published, few studies analyzing hereditary breast cancers exist. Small sample sizes are a common denominator of most of the early studies that have been published. High-quality RNA and DNA are a prerequisite for conducting microarray analysis, wherefore access to frozen tumor tissue is necessary. The small study numbers can be explained by the fact that hereditary breast cancers account for only a minor fraction of breast cancer cases and access to frozen tumor tissue is often limited. Recently, several studies on larger cohorts using newer generations of microarray platforms have been published. In the following, the early and the more recent studies will be discussed. Tables 1 and 2 represent overviews of the published transcriptome profiling studies and genomic profiling studies of hereditary breast cancers, respectively.
Published microarray RNA profiling studies of hereditary breast cancers. Numbers refer to number of BRCA1, BRCA2, non-BRCA1/2, and sporadic breast cancers primary tumors analyzed. Studies with sample overlap are listed together.
Published array-CGH studies of hereditary breast cancers. Numbers refer to number of BRCA1, BRCA2, non-BRCA1/2, and sporadic breast cancers primary tumors analyzed. Studies with sample overlap are listed together.
Gene-expression profiling of hereditary breast cancer
The early studies
The first microarray-based study of hereditary breast cancers was published by Hedenfalk et al in 2001. 94 With an underlying hypothesis that germline BRCA1/2 mutations have a profound impact on the gene-expression pattern, they analyzed tumors from BRCA1 (n = 7) and BRCA2 (n = 8) mutation carriers and sporadic tumors (n = 7). The authors identified 51 genes whose variation in expression best differentiated the three groups of cancers. Two different classification schemes were used for classification of BRCA1 and BRCA2, one for separating BRCA1 tumors from non-BRCA1 tumors and another for separating BRCA2 from non-BRCA2 tumors. All seven BRCA1 tumors were correctly classified, while one non-BRCA1 tumor was misclassified as a BRCA1 tumor. Further investigations revealed hypermethylation of the BRCA1 promoter in the single misclassified tumors. Distinguishing BRCA2 tumors from non-BRCA2 tumors were less successful, predicting 5 out of 8 BRCA2 tumors correctly and 13 out of 14 tumors without BRCA2 mutations correctly. Furthermore, they identified 176 genes with distinct expression between BRCA1 and BRCA2 tumors with genes involved in DNA repair and apoptosis pathways to be higher expressed in BRCA1 relative to BRCA2 tumors. The study served as a proof-of-concept study; however, concerns have been raised because of the small sample size and a lack of appropriate matching according to clinical parameters such as ER-status, known to have profound impact on the gene-expression pattern. 95 In later BRCA1 classification studies by van'tVeer et al and Lisowska et al, samples were matched according to ER-status prior to BRCA1 classification.96,97 Lisowska obtained only near-random classification while van't Veer achieved high accuracy (95%) when classifying 17 ER- BRCA1 tumors and 21 ER-sporadic tumors. Based on absolute correlation coefficients they identified 100 optimal marker genes for use in a leave-out one cross validation (LOOCV) classification algorithm. Again, promoter hypermethylation was demonstrated in a sporadic tumor classified as BRCA1-like. The main concern has been that the genes used for classification were identified using all samples, including also the left-out ones, wherefore the classification performance may be biased because of possible information leakage. Notably, no gene overlap was seen between the gene signatures identified by van'tVeer et al and Hedenfalk et al.
Only few years after their first pioneering study, Hedenfalk et al analyzed the gene-expression patterns of 15 primary tumors and 1 metastatic tumor from eight hereditary breast cancer families where no BRCA1/2 mutations could be detected (non-BRCA1/2). 98 Based on class discovery analysis, the authors were able to identify 2 distinct and homogenous subgroups among the 16 tumors. Sixty genes were found to be differentially expressed between the two subgroups. Of these, ribosomal-related genes were overrepresented. Notably, all families in which multiple family members were examined remained intact when divided into subgroups. The authors noted that these subgroupings could reflect different underlying genetic predispositions; however, they never validated their observation.
Molecular subtypes of hereditary breast cancer
A pioneering study in 2000 by Perou and colleagues was the first to show that breast cancers can be divided into subtypes distinguished by differences in their gene-expression profiles. 99 In subsequent studies, these observations have been repeated in larger sample series, and it is now established that at least four intrinsic molecular subtypes exist, designated basal-like, luminal A (lumA), luminal B (lumB), and HER2-enriched. These subtypes correspond broadly to histopathological characteristics and correlate to clinical outcome. Basal-like cancers are mostly high-grade and TN tumors (ER-negative, PR-negative, and HER2-negative), while HER2-enriched cancers often show amplification and high expression of the HER2 (ERBB2) gene and a series of genes located in the ERBB2 amplicon. Cancers of the luminal subtypes are ER-positive. In addition, lumA is low-grade and PR-positive tumor, while lumB is often high-grade cancer and to some extent PR-negative. The intrinsic subtypes are found to be highly conserved across different microarray platforms and across tumors from distinct ethnic populations.100–104
The first study to investigate molecular breast cancer subtypes in association with hereditary breast cancers was conducted by Waddell et al in 2010. 105 Their study group comprised BRCA1 (n = 19), BRCA2 (n = 30), and non-BRCA1/2 (n = 25) hereditary breast cancers. Subtype prediction by the PAM50 classifier revealed that 74% BRCA1 tumors were basal-like, 73% of BRCA2 tumors were luminal (equally distributed among lumA and lumB), and 52% of non-BRCA1/2 tumors were lumA. These observations has subsequently been confirmed, first by Jönsson and colleagues in a large cohort comprising BRCA1 (n = 34), BRCA2 (n = 39), and non-BRCA1/2 (n = 195) and more recently by our group in a sample series of 33 BRCA1, 22 BRCA2, and 70 non-BRCA1/2 samples.106,107 Strong associations between basal-like and BRCA1-associated breast cancers (85% and 61%), as well as lumB and BRCA2-associated cancers (56% and 73%) were observed in both studies. Fernández-Ramires et al analyzed 14 tumors from BRCA1 mutation carriers of which 9 were ER-negative. 108 In the study, they were able to substratify the ER-negative tumors into two groups with slight differences in the magnitude of the expression of immune response transcripts and REL/NFκB transcription factors. These subgroups showed some association with the BRCA1 mutation type (protein truncating versus missense).
In a recent study, we analyzed 70 non-BRCA1/2 cancers and found that the distribution of subtypes was markedly different from the distribution found among BRCA1/2 mutation carriers. 34 All five molecular subtypes were found within the non-BRCA1/2 tumor class. The majority of non-BRCA1/2 tumors were mainly classified as lumA (47%) or lumB (26%), while fewer were basal-like (13%), HER2-enriched (10%), and normal-like (4%). The distribution of molecular subtypes among the non-BRCA1/2 tumors was found to be similar to the distribution of sporadic tumors; although a tendency toward more non-BRCA1/2 tumors was basal-like while fewer were classified as lumB. These numbers were highly concordant with the previous studies.105,106 From 11 families, tumor material from more than one affected individual was included in the study. Surprisingly, we found that members of the same family shared the same tumor subtype in 8 of the 11 families. Three of the families were characterized by lumA tumors only (including the three-case family), three families had lumB tumors, one had HER2-enriched tumors, and one had only basal-like tumors. To confirm our observations, we subtyped the samples of Hedenfalk et al 98 consisting of tumors from a total of five high-risk families. The patterns of aggregation of molecular subtypes within families were confirmed in four of the families. These findings could indicate an underlying common genetic basis in these families. The family members may carry an inherited susceptibility not just to breast cancer but to a particular subtype of breast cancer. In support of the “same gene–-same subtype” hypothesis, in another study by Waddell et al, the authors noticed that all tumor biopsies from ATM mutation carriers included in their study were classified as luminal (four lumB and two lumA). 109 Within a large non-BRCA1/2 family, four out of five family members were classified as lumA. Furthermore, a study by Nagel et al included a group of 26 breast tumors from CHEK2*1100delC carriers; all were classified as luminal tumors (8 lumA and 18 lumB). 110 The cancer-risk and tumor subtype may either be a result of private mutations in high-penetrance genes or be a result of multiple low/moderate-penetrant genes acting in concert. In light of these findings, future genetic analysis may benefit from subgrouping families into molecularly homogeneous subtypes in order to search for new high-penetrance susceptibility genes.
In a study by Fernández-Ramires et al of 14 non-BRCA1/2 hereditary breast cancers, 2 subgroups very similar to the intrinsic lumA and lumB subtypes were identified. 111 By comparing the lumA non-BRCA1/2 with sporadic tumors of the same subtype, they identified a set of 157 deregulated genes of which 21 could be linked to DNA damage response canonical pathway. No differences between lumB non-BRCA1/2 and its sporadic counterpart were detected.
BRCA1/2 classification within tumor subgroups
Because of the strong association between BRCA1/2 mutation status and molecular subtypes, we choose to stratify tumor samples according to molecular subtype prior to classification in order to avoid potential confounding effects. 107 By conducting BRCA1-versus-sporadic classification within only basal-like samples using support vector machine (SVM)-based LOOCV, we found that basal-like BRCA1 tumors could successfully be distinguished from sporadic tumors of the basal-like subtype with high accuracy (balanced accuracy: 83%, sensitivity: 85%, specificity: 80%). Likewise, BRCA2 classification was performed among lumB tumors, as the vast majority of BRCA2 tumors were of the lumB subtype. This resulted in a balanced accuracy of 89% (sensitivity: 88%, specificity: 90%). We sought to validate our subtype-specific BRCA1/2 signatures in a set of independent samples. Using the data sets of van'tVeer and Jönsson studies,96,112 we validated our BRCA1/2 signatures. Using the two independent data sets, we were able to successfully validate both the BRCA1 consisting of 100 genes and BRCA2 signature of 110 genes with high accuracies (82–87%). Our results support the hypothesis that BRCA1-associated tumors represent a distinct biological subgroup among basal-like tumors, which has been a topic of debate. Likewise, BRCA2-associated tumors pose a distinct subgroup among lumB tumors. Next, we applied the subtype-specific signatures to predict BRCA1 and BRCA2 associations among non-BRCA1/2 tumors, respectively. 34 We found that seven out of nine basal non-BRCA1/2 samples were BRCA1-like. In a similar approach using our lumB BRCA2 signature, we identified 7 out of 18 (39%) lumB non-BRCA1/2 tumors to be BRCA2-like. This could indicate BRCA1/2 deficiencies in these tumors, either caused by an inactivating mutation not detected by current methods or epigenetic silencing such as promoter hypermethylation of the BRCA1/2 genes or other susceptibility genes in the same pathway. In three of the BRCA1-like tumors, we provided evidence for epigenetic inactivation of BRCA1 by promoter methylation.
Although additional validation studies are required, indication of BRCA1/2 involvement (BRCAness), using subtype-specific BRCA1/2 signatures in combination with subtype classification, RNA profiling could potentially be valuable as a tool for distinguishing pathogenic mutations from benign variants, for identifying undetected mutation carriers, and for selecting patients sensitive to new therapeutics such as PARP-inhibitors.
Genomic aberration in hereditary breast cancer
With the implementation of microarray-based comparative genomic hybridization (array-CGH), high resolution analysis of chromosomal aberrations in tumor samples became easy accessible. The first study utilizing array-CGH for analysis of hereditary breast cancers was in 2005 by Jönsson et al. 113 The investigators obtained genomic profiles from BRCA1 (n = 14), BRCA2 (n = 12), and sporadic (n = 26) breast cancer patients. Using SVMs classification, 11 of 12 samples with BRCA1 mutations were correctly identified in the BRCA1 classification, while 4 non-BRCA1 samples were misclassified. BRCA2 classification resulted in 9 of 12 samples correctly classified and 4 misclassified. In addition, they identified 4p, 4q, and 5q as frequently lost in BRCA1 tumors relative to sporadic tumors. 7p and 17q24 were found to be frequently gained in BRCA2 compared to sporadic tumors. The study observed highest frequencies of copy number alternations in BRCA1 tumors. The regions described as discriminative by Jönsson et al were further evaluated in an array-CGH study by Melchor et al in a series composed of BRCA1 (n = 19), BRCA2 (n = 24), non-BRCA1/2 (n = 31), and sporadic tumors (n = 19). 114 The authors observed that the regions mainly differentiated ER-positive tumors from ER-negative tumors rather than BRCA mutation status, caused by the fact that in the Jönsson study, all BRCA1 tumors were ER-negative and all BRCA2 tumors were ER-positive. On this background, it was suggested that ER-status should be considered in future study designs. Five years after their first study, Jönsson and colleagues reported in 2010 a study of a set of 346 primary tumor samples (plus 13 metastases), including BRCA1 (n = 17), BRCA2 (n = 31), non-BRCA1/2 familiar (n = 126), and sporadic (n = 172) tumors. 112 The study identified genomic subtypes by unsupervised clustering of the copy number profiles designated basal-complex, 17q12, luminal-complex, luminal-simple, amplifier, and mixed. The genomic subtypes were highly concordant to the intrinsic subtypes determined by gene-expression. The majority of BRCA1 tumors (77%) had the basal-complex subtype (comparable to basal-like), while the majority of BRCA2 (78%) were luminal-complex (comparable to lumB). The familial non-BRCA1/2 samples were distributed across the different genomic subtypes similar to the sporadic cancers. Luminal-complex BRCA2 tumors were characterized by losses on 3p21.31, 3p14.1, 6q16.2, 13q14.2, 14q24.3, and 22q13.31 and gains on 17q25.3 compared with non-BRCA2 tumors in the same genomic subtype, whereas the sporadic tumors showed more-frequent gain of 11q13.3. No region was found to differ significantly between BRCA1 and non-BRCA1 tumors in the basal-complex subtype.
From these array-CGH studies, among others, it also became clear that patterns of genomic aberrations are highly influenced by the ER/HER2 status and the molecular subtype of the tumors. To construct subtype-independent BRCA1 and BRCA2 classifiers, Joosse and colleagues used ER-matched tumor samples for feature selection and training of the classifiers. In Joosse et al, 115 18 BRCA1 and 32 sporadic tumors were used as training set for construction of a BRCA1 classifier, while 16 BRCA1 and 16 sporadic tumors made up the test set. Using this approach, a sensitivity of 88% and a specificity of 94% when applied to the test set were obtained. The classifier was also used to identify BRCA1-like tumor profiles among 48 non-BRCA1/2 tumors from HBOC families. The results showed that 2 of the 48 non-BRCA1/2 breast tumors exhibited chromosomal aberrations similar to those found in BRCA1-mutated tumors. Further analysis demonstrated LOH of BRCA1 in both cases and hypermethylation of BRCA1 gene promoter in one case. The most abundant genomic abbreviations that differed between BRCA1 and sporadic tumors were 3q22-27 (gain), 5q12-14 (loss), 6p23-22 (gain), 12p13 (gain), 12q21-23 (loss), and 13q31-34 (gain). A similar approach was used by Joosse et al for construction of a BRCA2 classifier, using a training set of 28 BRCA2 and 28 sporadic tumors. 116 From a validation set of 19 BRCA2 and 19 sporadic tumors, they achieved a sensitivity of 89% and a specificity of 84%. Testing a set of 89 cases from non-BRCA1/2 high-risk families, 12 were found to exhibit a high level of similarity with true BRCA2-mutated breast tumors. In three cases, additional indications of dysfunctional BRCA2 were found by determining allele-specific mRNA expression. Nine other cases demonstrated LOH/allelic imbalance of the BRCA2 locus, indicating possible loss of BRCA2 not detected by standard diagnostic procedures. Chromosomal aberrations specific for BRCA2-mutated tumors were loss of 13q and 14q and gain of 17q.
Conclusion and Future Perspectives
The results from the last decade of pathological and molecular characterization of hereditary breast cancer have unquestionably contributed with important insights into the biological mechanism underlying hereditary breast cancers. It is now well established that tumors of hereditary breast cancers are not phenotypically distinct groups of cancers, instead they are associated with the intrinsic molecular subtypes. BRCA1 tumors are mainly TN/basal-like, BRCA2 tumors are ER+/lumB cancers, and non-BRCA1/2 tumors are more phenotypically heterogeneous but most often of the ER+/luminal subtypes. The described studies also stress the importance of careful study design. Because of the strong association to the molecular subtypes, proper sample matching is important to avoid bias in order to detect genomic features unique for hereditary breast cancers. By stratifying for ER-status or molecular subtypes, recent RNA- and DNA-based classification studies have demonstrated that tumors from BRCA1 and BRCA2 mutation carriers represent distinct biological entities among ER–/basal-like and ER+/lumB tumors, respectively. Signatures based on transcriptome as well as genome profiling have proven suitable for prediction of BRCA1/2 status with high accuracies and have been shown to have the capacity to identify tumors with BRCA1/2-like molecular phenotypes among tumors with no recognized BRCA1/2 mutation. Although more research on larger cohorts is required, molecular signatures could have the potential to improve diagnostics by facilitating the clinical interpretation of the large number of sequence variants of unknown clinical significance found in the BRCA1/2 genes by distinguishing pathogenic mutations from benign variants. Such signatures could also be used as a tool for preselecting patients for mutation screening, as a significant proportion of BRCA1 and BRCA2 germline mutation carriers do not have a family history of breast cancers. New targeted therapies such as PARP-inhibitors have been demonstrated to be effective treatments for BRCA1/2 mutation carriers because of dysfunctional HR DNA repair. In addition to germline mutations, other mechanisms, such as somatic and epigenetic inactivation of BRCA1/2, can lead to BRCA-deficiency and impaired HR DNA repair. Molecular signatures could potentially prove to provide a general method for detecting BRCA-deficient tumors sensitive to new target therapies making it applicable for optimal treatment decisions. Molecular profiling may also be valuable to future genetic analysis by stratifying tumor/families into molecularly homogenous subgroups to aid the search for new breast cancer susceptibility genes. The landscape of hereditary breast cancer is starting to emerge; however, studies of non-coding RNA expression (such as microRNA and lncRNA), NGS, as well as epigenetic studies will undoubtedly add important details to the description of the complex genetic architecture underlying hereditary breast cancer.
Author Contributions
Wrote the first draft of the manuscript: MJL. Contributed to the writing of the manuscript: MJL, MT, AMG, TK. Agree with manuscript results and conclusions: MJL, MT, AMG, TK. Jointly developed the structure and arguments for the paper: MJL, MT, AMG, TK. Made critical revisions and approved final version: MJL, MT, AMG, TK. All authors reviewed and approved of the final manuscript.