
Abstract
Motivation
Despite more than two decades of research, HIV resistance to drugs remains a serious obstacle in developing efficient AIDS treatments. Several computational methods have been developed to predict resistance level from the sequence of viral proteins such as reverse transcriptase (RT) or protease. These methods, while powerful and accurate, give very little insight into the molecular interactions that underly acquisition of drug resistance/hypersusceptibility. Here, we attempt at filling this gap by using our Monte Carlo feature selection and interdependency discovery method (MCFS-ID) to elucidate molecular interaction networks that characterize viral strains with altered drug resistance levels.
Results
We analyzed a number of HIV-1 RT sequences annotated with drug resistance level using the MCFS-ID method. This let us expound interdependency networks that characterize change of drug resistance to six selected RT inhibitors: Abacavir, Lamivudine, Stavudine, Zidovudine, Tenofovir and Nevirapine. The networks consider interdependencies at the level of physicochemical properties of mutating amino acids, eg,: polarity. We mapped each network on the 3D structure of RT in attempt to understand the molecular meaning of interacting pairs. The discovered interactions describe several known drug resistance mechanisms and, importantly, some previously unidentified ones. Our approach can be easily applied to a whole range of problems from the domain of protein engineering.
Availability
A portable Java implementation of our MCFS-ID method is freely available for academic users and can be obtained at: http://www.ipipan.eu/staff/m.draminski/software.htm.
Introduction
For more than two decades rapid emergence of drug- resistant mutants remains a serious obstacle to designing efficient anti-HIV therapies. Approximately 10 9 new viral particles are produced in a single infected individual every day and 3 × 105 mutations emerge in this population. 1 Some mutations lead to the production of functionally impaired viruses whereas other give rise to drug-resistant strains. Clinical studies have demonstrated that after 2–4 weeks of treatment with anti-HIV drugs the wild-type virus is almost completely replaced by drug-resistant strains. 2
Currently, 25 different anti-HIV drugs are approved for treatment and many new drug candidates have reached phase I and phase II clinical trials. Twelve drugs belong to a group of RT inhibitors targeted against an important viral enzyme, reverse transcriptase (RT). 3 Provided with a single-stranded viral genome RNA template, the RT catalyzes the synthesis of double-stranded proviral DNA. In turn, the proviral DNA is incorporated into a host genome, expressed and replicated by the native cellular machinery. This results in the production of new HIV particles capable of infecting other host cells and, thus, completing the HIV life cycle.
Two groups of reverse transcriptase inhibitors are in clinical use, namely the nucleoside/-tide RT inhibitors (NRTIs) and the non-nucleoside RT inhibitors (NNRTIs). NRTIs mimic dNTPs, the natural substrates of the RT enzyme, but once incorporated into a newly synthesized strand, they terminate DNA elongation. The members of the second group, the NNRTIs inhibit RT by binding in the so-called NNRTI-binding pocket which, in turn, induces function-disrupting structural changes in the enzyme. The state-of-the-art highly-active antiretroviral therapy (HAART) regimens frequently include a nucleoside RT inhibitor, a non-nucleoside RT inhibitor and a protease inhibitor. 4
A need for new antiviral drugs has been recognized. New generation of drugs should be effective against viruses that carry the already known resistance mutations. Drugs belonging to the so-called “second generation RT inhibitors”, eg, Dapivirine, have been introduced recently. 5
The availability of resistance-annotated sequence data and the inherent complexity of analyzing the 560 amino acids of the RT p66 subunit call for an application of suitable computational methods. In our previous study, 6 we have developed a number of rule-based models that relate mutation-induced changes in the physicochemical properties of HIV-1 RT to the level of resistance to several antiviral drugs. A remaining issue was how the individual mutations interact in altering resistance.
Approach
We used our interdependency discovery method based on Monte Carlo feature selection (MCFS-ID)7,8 to reveal molecular interaction networks leading to drug resistance. It results in a network of resistance- altering molecular interactions between physicochemical properties of mutating amino acids. We applied the method to find molecular interaction networks that underly resistance to six anti-HIV drugs: four nucleoside RT inhibitors (Abacavir, Lamivudine, Stavudine, Zidovudine); one nucleotide RT inhibitor (Tenofovir) and one non-nucleoside RT inhibitor (Nevirapine).
The molecular-interaction networks that we constructed are based on thousands of decision-tree classifiers. Each pair of immediately neighboring nodes reflects their joint, statistically valid contribution to co-determination of the outcome. We used this fact to elucidate molecular interaction networks between mutating amino acids.
Methods
Data and discretization
From the Stanford HIV Database 9 we obtained a number of resistance-annotated amino acid sequences of HIV-1 reverse transcriptase p66 subunit. The 91% of the sequences were complete within the first 240 aa sites, 31% within all the sites. As suggested by Zhang et al, 10 we used only the sequences with resistance determined by the Monograms® PhenoSense® assay. Following well established clinical practice, we discretized continuous drug resistance values into three classes: “susceptible”, “moderately resistant” and “resistant”. Cut-off values used for the discretization were taken from the work by Rhee et al. 9
Description
Rather than modeling the resistance in terms of muting amino acids (eg, work by Beerenwinkel et al 11 ), we represent mutations as changes in the physicochemical properties of the enzyme. In contrast to the approach proposed by Kjaer et al. 12 we consequently use the same set of descriptors for each class of antiviral drugs. Previously,13,6 we have selected 7 descriptors–physicochemical properties from the aaIndex database; 14 see Table 1. The descriptors are relatively low-correlated (cf. Kierczak et al 6 ), easy to interpret by structural biologists and cover various aspects of protein biophysics including solvent interactions, thermodynamics, secondary and tertiary structure and electrostatic properties. We use the selected properties to describe each amino acid in the sequence.
Descriptors of the physicochemical properties of amino acids used in this study.

Thus, each sequence is described by 3,920 features (560 aa × 7 physicochemical properties). Each amino acid in the sequence was encoded as a difference between the 7-vector of its properties and the 7-vector representing properties of the corresponding site in the wild type HXB2 reference sequence. Therefore, if no mutation was observed at a given position, it was encoded by the zero vector. Our final dataset is an ensemble of sequences described in such way and annotated with their resistance category. The detailed characteristics of each data set are provided in Table 2.
The detailed characteristics of the analyzed datasets. Table summarizes the exact numbers of examples per drug and per resistance class.

Monte Carlo feature selection and interdependency discovery
The description step was followed by the selection of significant resistance-altering interdependencies between the physicochemical properties of the 560 amino acids of p66. We applied Monte Carlo Feature Selection (MCFS) method as described in Dramiński et al. 7 In our most recent study, 22 we show that, unlike Breiman's Random Forests, our method is not biased towards features with many categories. The MCFS method relies on the construction of a large number of decision trees. We trained the trees on s different random subsets of attributes and different random subsets of objects. For each subset, we constructed t trees. Each of the trees was trained and evaluated on a different, randomly selected training set–test set pair. The evaluation results obtained from all the s · t trees let us build a ranking of physicochemical properties reflecting their influence on drug-resistance. The final ranking of all features does not give the information about the cut-off value that separates informative features from the non- informative ones. We find the cut-off by applying Student's t-test with its critical value (significance level set to: P-value ≤ 0.05) determined on a set of rankings that have been obtained for data sets with randomly permuted values/labels of the class attribute. For the detailed description and for the comparison of the MCFS method to other feature selection techniques see our previous works.6–8
Our approach to interdependency discovery (ID) is significantly different from known approaches which consist in finding correlations between features or finding groups of features that behave in some sense similarly across samples. A typical bioinformatics example of this problem is finding co-regulated features, most often genes or, rather more precisely, their expression profiles. Searching groups of similar features is usually done with the help of various clustering techniques, frequently specially tailored to a task at hand.23–26
In our approach to interdependency discovery (ID) we focus on identifying features that cooperate in determining that a sample belongs to a particular class. Assume that for a given training set of samples, a rule-based classifier (or a decision tree), has been constructed. Now, for each class, a set of decision rules defining this class is provided. Each decision rule is in fact a conjunction of conditions imposed on a particular subset of separate features and, thus, points to some interdependencies between these features. It is worthwhile to note that in this way any nonlinear interdependencies are sought which best correlate with the decision attribute (within regression setting such an approach can be said to rely on additive expansion interaction models based on regression trees 27 (Sec. 2)).
This general idea of looking at interdependencies among features seems to be rather plausible but we have refined it further, so that it is not dependent on just one classifier. In each of the s · t trees the nodes represent features (a given node represents the feature on which the split is made). For each path in a tree, we define the distance between two nodes as the number of edges between them. Now, the strength of interdependency between two nodes along the same path in the tree is defined as the inverse of the distance between them. For a given pair of features, the strength of interdependency between them is computed as the sum of the strengths between them along all paths in all trees (if the two features do not appear along a path, the contribution of this path to the overall strength is zero). Here, we consider only the pairs where the distance d ≤ 3. This value is a trade-off between the complexity of reviewing a tree and searching for the strongest dependencies. Eventually, we normalize every strength by dividing its value by the strength-value of the strongest observed interdependency.
Since the strength is calculated on the basis of thousands of trees rather than on the basis of just a single classifier, it provides stable and reliable information about the degree of the observed interdependency between the features.
Final results of the analysis are presented in the form of graphs where nodes represent features and edges represent interdependencies. Only the nodes corresponding to the informative features are shown. An edge connecting two nodes corresponds to the observed interaction between them. The interaction relation is not transitive, ie, if node A interacts with node B and node B interacts with node C, it does not imply that node A interacts with node C. It is, however, plausible that the adjacent pairs of interacting nodes co-determine resistance. Nodes corresponding to the same aa site occupy the same level and follow top to bottom order from N-terminal to C-terminal part of the sequence.
We visualize the discovered networks in Figure 1. Each node represents a physicochemical property at a particular site. Color of a node corresponds to the property and the number to the position of the site in the RT sequence. For instance (Fig. 1a, leftmost part), free energy of solution in water at site 184 interacts frequently with either of the following site-property pairs: normalized frequency of turn at site 41, isoelectric point at site 41, normalized frequency of turn at site 210 or transfer free-energy from octanol to water at site 215. These interactions determine the level of resistance to Abacavir. Interestingly, all properties but polarity appear in the resistance-to-Abacavir network at site 184 (Fig. 1a–d). This indicates high degree of conservation at the site and points out its central role in resistance complexes.
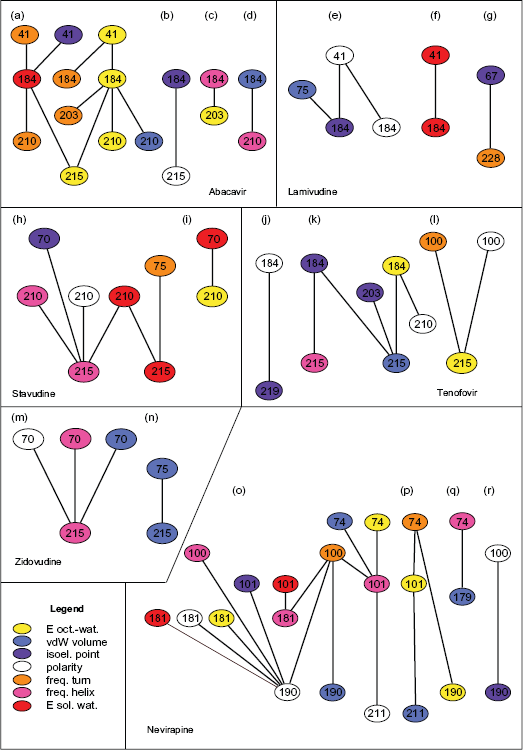
Interaction networks determining resistance to six HIV-1 RT inhibitors. Numbers refer to the aa sites of the RT p66 subunit. Colors correspond to physicochemical properties.
Discussion
We examined the top 20% of interactions among the physicochemical properties that contribute to resistance and analyzed these networks in attempt at better understanding the combined contribution of individual mutation-sites. The discovered networks are presented in Figure 1.
Below we provide a brief overview of the RT structure and function. Next, we describe structural properties of each of the 17 aa residues that occur in the discovered networks. This is followed by an individual discussion of each drug-resistance network. A summary of the findings concludes this section.
The RT is responsible for both RNA-dependent and DNA-dependent DNA polymerization. The enzyme consists of two subunits: 560 aa-long p66 and 440 aa-long p51. Although both the subunits share 440 amino acids, their arrangement and function differ significantly: the functional p66 subunit contains a DNA-binding groove and the active site whereas p51 lacks enzymatic activity and serves as a scaffold for p66. Mutations in the p66 subunit are responsible for the emergence of drug resistance. The ternary structure of the p66 subunit is often compared to a right hand consisting of a thumb, a palm and a fingers subdomain.2,5 These three, together with the connection subdomain, are present in both the p55 and the p66 subunits. The p66 subunit contains also an RNaseH domain.
The mechanism of polymerization by HIV-1 RT is similar to that described for other DNA polymerases and involves: 1) template/primer binding to the enzyme; 2) binding of dNTP and bivalent cations (Mg2+) to the RT-template/primer complex; 3) formation of a phosphodiester bond between the 3′-OH primer terminus and the α-phosphate of a dNTP; 4) translocation of elongated DNA primer from the dNTP binding site to the primer site or release of the template/primer complex. A large conformational shift and rotation of the fingers subdomain towards the thumb subdomain occurs upon step 2.28,29
The palm subdomain contains three catalytically active residues: Asp 110, Asp 185 and Asp 186 embedded in a hydrophobic region.29,30 Both Asp 185 and Asp 186, together with Tyr 183 and Met184 constitute a highly-conserved YXDD motif found in all reverse transcriptases. 2
It has been suggested that during polymerization the fingers subdomain and both alpha-helices of the thumb form a clamp that holds the nucleic acid in the right place over the palm. The template/primer interactions occur between the sugar-phosphate backbone of the DNA/RNA and residues of the p66 subunit.2,28,29
Seventeen different amino acid residues occur in the discovered interaction networks (Figs. 1 and 2): Met 41, Asp 67, Lys 70, Leu 74, Val 75, Leu 100, Lys 101, Val 179, Tyr 181, Met 184, Gly 190, Glu 203, Leu 210, Arg 211, Thr 215, Lys 219 and Leu 228. Mutations at residues 100, 101, 179, 181, 190, 203 and 228 have been associated with resistance to NNRTI drugs.9,31 Mutations at residue 211 can be found in some non-B HIV subtypes 32 and they are suspected to increase viral fitness by alleviating the deleterious effect of other drug-resistance mutations. 33 Mutations at the remaining sites are known to contribute to resistance to NRTI drugs.9,3,31
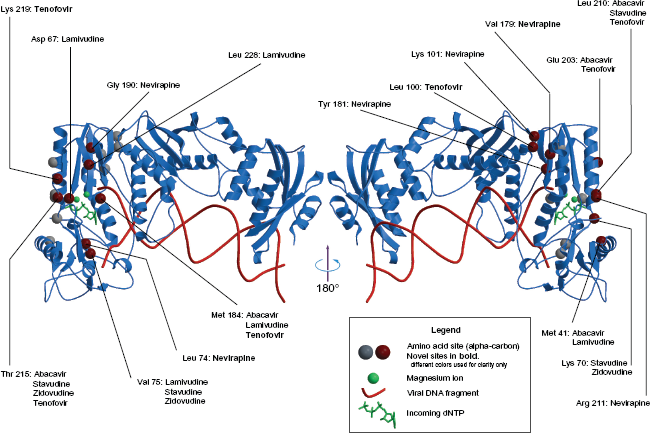
Resistance to RT-inhibitors. (PDB structure: 1RTD). A number of amino acid sites that occur in the resistance networks is mapped onto the 3D structure of the HIV-1 RT p66 subunit. For more clarity the structure to the right is rotated 180° Novel sites that have not been associated with resistance to a particular drug are presented in
Methionine at residue 184 is located in the close vicinity of the catalytic triad and may therefore play crucial role in modifying RT selectivity towards incoming dNTPs and drug triphosphates (drug-TP). Residues Met 41 (inside the 28–44 alpha-helix), Leu 74 and Val 75 belong to the fingers subdomain, part of which is involved in the template positioning. 34 Residues Asp 67 and Lys 70 belong to the fingers domain while residues Leu 210 and Thr 215 are embedded inside the palm subdomain and constitute a fragment of the dNTP binding pocket. 29 They are located opposite to Met 184. Asp 67 remains in direct contact with a Mg2+ ion that catalyses enzymatic reaction. 30 Glu 203, Leu 210 and Arg 211 are all part of the 195–212 alpha-helix. The C-terminal part of this helix interacts directly with an incoming dNTP or drug-TP. Leucine 228 is a part of a short (227–229) beta-strand in the palm subdomain. Leucine 100 is located in the palm, on the surface of the NNRTI-binding pocket. Mutations of Leu 100 give moderate resistance to a number of NNRTI drugs. 5 Tyrosine residues 181 and 188 are involved in the ring stacking interaction with NNRTIs and their mutations to non-aromatic amino acids leads to drug resistance. 5 Residues Phe 227, Leu 228 and Trp 229 are building-blocks of the NNRTI-binding pocket. Mutations resulting in the decrease of chain-bulk at residue 227 lead to loss of favorable contacts with some NNRTIs. 35 Tryptophane 229 apperas to be highly conserved and it has been suggested that new generation of drugs should interact with this particular residue in order to avoid drug resistance. 36
Figure 1 presents all the discovered networks that we discuss below. Whenever we refer to a graph by a letter eg, (a)—we refer to this figure. The following section discusses each RT inhibitor individually. In supplementary materials, Figure S1, we present the detailed information on the distances between α-carbon atoms of interdependent and non-interdependent significant residues.
Abacavir
Five different amino acid sites: 41, 184, 203, 210 and 215 are selected by the MCFS-ID method (a)–(d). In graphs: (b), (c) and (d), either of the 203, 210 and 215 residues, interacts with residue 184. Each of these interactions appears to be a good descriptor of the dNTP binding pocket which follows directly from the 3D structure of the enzyme, see eg, work by Menedez-Arias 29 and Figure 2. In graph (a), residue 184 co-occurs with two sites: 41 and 203. Since Glu 203 and Leu 210 are parts of the same alpha-helix, mutations at site 203 may affect structure and position of this helix. Therefore the 184–203 interaction may be another good descriptor of the selectivity of the pocket. This is further supported by the fact that the normalized frequency of alpha-helix has been selected at site 203. The second pair (41–184) reflects the possible interaction between the binding pocket and the fingers subdomain.
Lamivudine
Graphs (e)–(g), present interactions discovered for Lamivudine. Apart from the already discussed pairs, isoelectric point at site 67 and normalized frequency of turn at site 228 constitute a pair. Closer look at the 3D structure (Fig. 2) of the p66 subunit reveals that Leu 228 is located inside the short (227–229) beta sheet. Mutations disturbing this structure may result in repositioning the 178–183 and the 186–191strand. This, accompanied by mutations at residue 67, may affect substrate selectivity of the enzyme preventing incorporation of drug-triphopsphates.
Stavudine
A new type of interaction emerges in the case of Stavudine, another NRTI drug ((h), (i)). Here, normalized frequency of turn at residue 75 (the 71–75 strand) co-occurs with free energy of solution in water at residue 215; see graph (h). The latter descriptor characterizes hydrophobic properties of amino acid and its emergence in the context of the hydrophobic dNTP-binding pocket is not surprising. Changes in both the electrostatic properties and the propensity to form alpha-helix characterize drug-resistance mutations at site 210. Interestingly enough, an interaction involving hydrophobicity-related properties at sites 70 and 210 has been selected by the MCFS-ID; see graph (i).
Tenofovir
In the case of Tenofovir (graphs (j)–(l)), a NRTI drug, normalized frequency of turn is selected at site 100. Mutations at this particular site are typically connected with resistance to NNRTI drugs only.5,3,31 It might have emerged as a result of past treatment, but it may also play a role in resistance to Tenofovir. Although its significance in the context of NRTI drugs remains unclear, this in silico discovered interaction deserves further attention. The two remaining groups of interactions (graphs (k) and (l)) always involve site 184 and either of the sites located within or near the 195–212 alpha-helix. These interactions seem to characterize geometry and general properties of the dNTP-binding pocket.
Zidovudine
Interaction of sites 75 and 215 is also important for resistance to Zidovudine (graphs (m) and (n)). Here, mutations that lead to the change of van der Waals volume seem to matter (Fig. 2). Site 215 is also involved in interactions with site 70; see graph (m). Both sites are also parts of the dNTP binding pocket.
Nevirapine
Several interactions selected as relevant in the case of Nevirapine (graphs (o)–(r)) seem to reflect properties of the NNRTI-binding pocket. Mutations at sites 100, 101, 181 and 190 are well known to contribute to resistance.3,31 The selected interactions involve also site 74 that is associated with resistance to NRTI drugs, but not to NNRTIs. The occurrence of the 74 site in this context may, again, be the result of past treatment. The MCFS-ID selects also sites 179 and 211. Valine 179 is located in the NNRTI-binding pocket and mutations there have been associated with drug-resistance. 5 Our analysis indicates that changes in normalized van der Waals volume play important role in the emergence of resistance. Mutations at site 179 seem to interact with NRTI mutations at site 74 (graph (q)). Although mutations at site 211 have not been reported as contributing to resistance, mutations at the adjacent site 210 are well known. It is normalized van der Waals volume that has been selected at this position. We can speculate that certain mutations at site 211 disrupt the structure of the 195–212 alpha-helix thus contributing to resistance.
Conclusion
We were able to uncover and analyze several networks of molecular interactions responsible for the change of resistance levels to six antiviral drugs. The discovered networks encompass numerous previously known resistance mechanisms which is a strong evidence of the validity of the method. Significantly, application of the MCFS-ID method pinpoints also some new mechanisms potentially affecting drug resistance levels. These newly discovered mechanisms deserve further experimental investigation. While mechanisms leading to resistance to Lamivudine and Zidovudine are described by relatively simple networks, resistance to Abacavir and Nevirapine appears to be determined in more complex way. There are several structural fragments and sites that seem to play central role in altering resistance. For instance, site 184 plays important role in resistance to Abacavir, Lamivudine and Tenofovir. Similarily, many members of the 195–212 helix appear in the discovered networks indicating its functional importance. On the other hand, there are interactions and sites that emerged in the context of only one drug eg, 67–228 interaction determining resistance to Lamivudine. Some networks eg, (l), (o)–(q) contain sites that were associated with resistance to another class of drugs. These may be the result of past treatments, but they may also play accessory role that, since difficult to notify, was previously neglected. It would be highly interesting to include information on the history of treatment and on viral fitness to our analysis. Unfortunately, there is not enough publicly available data containing this information. Last but not least, some of the discovered interactions may play compensatory role by increasing viral fitness. The novel interactions deserve further attention and laboratory validation.
Importantly, the method itself shows interactions between physicochemical properties of mutating amino acids. This gives deeper insight into molecular mechanisms of drug resistance. The majority of the drug resistance mutations act simultaneously, in cooperative manner. The MCFS-ID method allows us to see these complex interactions. In our opinion the presented approach can be applied to several similar problems in the domain of proteomics.
Footnotes
Supplementary Figure
Acknowledgement
The presented research has been partially supported by the Knut and Alice Wallenberg Foundation, by the Swedish Foundation for Strategic Research and by grant nr. N301 239536 from the Polish Ministry of Science and Higher Education.
The manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.