
Abstract
Introduction
Malaria, a potentially lethal mosquito-borne disease, has existed as a public health burden for many decades, currently placing 1.2 billion of the world's population at high risk.
1
This disease is caused by parasites belonging to
In addition to the adaptive changes brought about by the vast reorganization of cellular processes, the parasite has also the capability to remodel host cells, particularly erythrocytes, to suit its niche during infection. During a blood meal of a mosquito vector, motile infective form of parasite known as sporozoites are injected into the blood stream, which then travel to liver where they undergo rapid multiplication and differentiation to generate large number of merozoites. The blood stages of infection begin once these merozoites released from liver cells invade the erythrocytes. Throughout the intraerythrocytic stages of the parasite's life cycle, including ring, trophozoite, and schizont, the parasite establishes intricate mechanisms to remodel erythrocytes for its growth and survival.
3
Exploitative mechanisms achieved by the parasite include the acquisition of nutrients from the cytosol of red blood cells (RBCs) and from extracellular environ, mediation of cellular adhesion of infected RBCs to avoid splenic clearance, evading host immune response by associating anti-genically variant proteins with erythrocyte surface, and the establishment of protein-trafficking machinery. Much of these mechanisms can be attributed to the parasite proteins targeted to RBC membrane.4,5 The intricateness in the massive remodeling of host RBCs induced by the parasite and the unusual plasticity of the parasite's metabolism through the various stages of its life cycle has been well studied.3–6 Moreover, the characterization of
Over the past several years, substantial efforts have been made toward the development of computational framework to predict protein-protein interactions with the help of evolutionary information9,10 that are primarily based on experimentally known interactions documented in various databases. However, interactions identified based on homology alone need rigorous evaluation in order to filter interactions in biological context. Functionally relevant interactions can be systematically identified by the use of molecular details of three-dimensional (3-D) structures of protein-protein complexes. By virtue of similarity to the structure of a protein complex, it is possible to determine and assess putative interacting residues in the homologous protein pair based on conservation. The credibility of the predicted interactions can then be enhanced by integrating additional information such as gene expression and subcellular localization in order to assess their ability to bind physically in the pathological context. The significance of such structure-influenced transfer of interactions between organisms has been realized and has formed the basis of several frameworks.11–13 On similar grounds, computational efforts to predict protein-protein interactions across human and pathogen(s) of interest have been successfully achieved by many groups14,15 as well as by one of our groups earlier.16–19
The availability of completely sequenced genome of human and
Methodology
Datasets considered
Protein sequences of 5542 gene products of
To pursue structure-influenced recognition of protein interactions, two datasets were used: (i) a cumulative dataset of structures of transient protein-protein complexes published earlier27–29 and (ii) a dataset of domain-centric protein-protein interactions from Protein Family Interactions (iPfam) database. 30 iPfam database provides a comprehensive resource of domain-domain interactions that are formulated using combined information from the structures of protein complexes in Protein Data Bank 31 and their constituent sequence domains acquired from Protein Family (Pfam) database. 32 Since the current definitions are primarily based on the calculations performed on asymmetric unit of protein complexes, we have restricted our dataset specifically to those interaction definitions where the crystal asymmetric unit of a protein complex corresponds to a whole biological assembly. For the current analysis, we have considered heterodomain interactions from iPfam, ie, interactions between different protein domain families.
Generation of initial host-parasite interaction dataset
The identification of host and parasite proteins homologous to a known pair of interacting proteins forms the primary step in the recognition of host-parasite protein-protein interactions.
In order to determine homologs of structures of transient protein–-protein complexes, we used family specific structural identifiers catalogued in the database of Structural Classification of Proteins (extended version SCOPe 2.04).
33
SCOP, an extensive database of manually curated protein structural relationships, hierarchically classifies protein domains into class-fold-superfamily-family based on structural and evolutionary relationships. A family specific identifier or SCOP code for a structural domain holds the information pertaining to its corresponding classification. Since evolutionarily related proteins tend to exhibit interactions in a similar manner, the family specific SCOP identifiers retrieved for transient protein–-protein complexes were mapped to those identified in RBC and parasite proteins. The identification of SCOP domains in RBC and parasite proteins involved a search against a database of profile hidden Markov models
34
(HMMs) representing domains in proteins of known structure, at an
Filter 1: Refining the template dataset
iPfam attributes the terms intrachain and/or interchain interactions to domains in a protein complex on the basis of the nature of polypeptide(s) and their proximity in the 3-D structures. We excluded the intrachain heterodomain interactions retrieved from iPfam, which were mapped to a single host RBC or a single parasite protein, as such interactions are less likely to occur across species. We had observed that homologs of co-occurring heterodomains in a multidomain protein rarely correspond to two different interacting proteins. 37 Considering this established observation, the exclusion of intrachain heterodomain interactions from the template dataset minimized the occurrence of false-positive predictions. Protein–-protein complexes that constituted synthetic constructs were also eliminated. All the protein complexes and the putative RBC–-parasite protein–-protein interactions deduced were manually curated to ensure their biological relevance.
Filter 2: Pruning intrahost interactions
Similar to the pruning steps in our previously published study, 19 we did not consider intrahost and intrapathogen interactions in our subsequent analyses. These interactions usually correspond to ubiquitous interactions that are conserved within most organisms. We also eliminated those RBC-parasite interactions where the RBC proteins are also capable of exhibiting intrahost interactions. In other words, when the interfacial region of an intrahost protein–-protein interaction is comparable to that of a host-parasite interaction, by virtue of similarity of host and parasite proteins to a single template protein complex, the predicted RBC-para-site interactions are not considered. This step ensured the recognition of targetable host–-parasite protein–-protein complexes. iPfam attributes the terms intrachain and/or interchain interactions to domains in a protein complex on the basis of the nature of polypeptide(s) and their proximity in the 3-D structures. We excluded intrachain heterodomain interactions retrieved from iPfam, which were mapped to a single host RBC protein.
Filter 3: Integrating additional information to extract biologically feasible interactions
Expression profile of parasite proteins for merozoite, ring, trophozoite, and schizont stages was extracted from PlasmoDB and mainly from three studies published earlier.38–40 Information on subcellular localization of parasite proteins was obtained from diverse sources. The parasite proteins that have been reported to constitute a host-targeting signal, ie, HT motif or PEXEL motif,41,42 and the exported proteins reportedly lacking PEXEL/HT motif (PNEPs)
43
were picked up. This criterion is of primary importance in recognizing feasible protein-protein interactions across
The subcellular localization data for RBC proteins (membrane, cytoskeleton, or cytosolic) were obtained from UniProt. This information becomes notably crucial in the RBC-
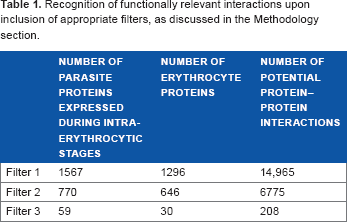
Given the set of 5542 parasite proteins and 1672 eryth-rocyte proteins, the number of possible interactions across the host erythrocyte and the parasite proteins is tremendously large. The use of appropriate filters at various levels, as discussed above, reduces false-positive predictions, thereby resulting in the recognition of probable host–-parasite interactions in the endogenous context. Table 1 outlines the number of protein–-protein interactions in the initial stages and the reduction in the number of false positives upon the inclusion of filters.
Recognition of functionally relevant interactions upon inclusion of appropriate filters, as discussed in the Methodology section.
A schematic representation of the protocol followed is shown in Figure 1.

Workflow. A schematic diagram of the steps taken to generate host-parasite protein-protein interaction dataset is shown. The steps start with the consideration of two datasets of transient protein-protein complexes, followed by the identification of their homologs in host and the parasite proteome. The encircled numbers correspond to equivalent subsections on filters in the Methodology section, which facilitated the recognition of biologically relevant host-parasite protein-protein interactions.
Results and Discussion
Probable host-parasite interactions and their influence on biological processes
The structure-influenced predictions in concert with a series of filters facilitated recognition of 208 physicochemically viable interactions accomplished by 59
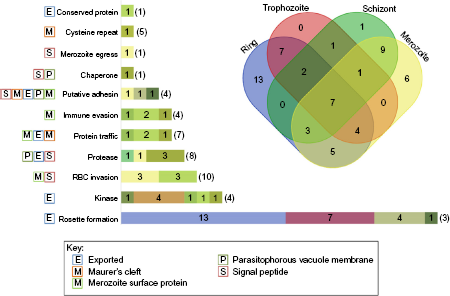
A schematic summary of RBC-parasite interactions. The stage-specific protein expression profiles of the parasite proteins are illustrated in the Venn diagram, while the bar graph depicts the number of parasite proteins under each functional category predicted to interact with host RBC proteins (denoted in brackets). The color codes in each bar are in correspondence with the color of stage-specific expression subsets shown in the Venn diagram. The single letter tags for all the functional categories of the parasite provide information on subcellular localization. The Venn diagram was created using an online tool (http://bioinformatics.psb.ugent.be/webtools/Venn/).
The potential influence on pathways and processes in RBC and the parasite were investigated based on the biologically feasible protein-protein interactions predicted across RBC and parasite proteomes. Functional annotations of the parasite proteins were obtained from PlasmoDB and the Malaria Metabolic Pathways database
47
and those of RBC proteins were retrieved from UniProt database. The putatively interacting protein pairs across the parasite proteins and the host RBC could be segregated into 11 and 10 functional categories, respectively, on the basis of the nature of their biological processes. Figure 2 illustrates the intraerythrocytic stage-specific distribution of 59
In addition to the host-parasite interactions potentially brought about by parasitic proteins belonging to nine functional categories (rosette formation, kinase, RBC invasion, protease, protein traffic, immune evasion, adhesion, chap-erone, and merozoite egress), we could identify two parasite conserved proteins of unknown function (PF3D7_0911300 and PF3D7_1463900), one of which is a cysteine repeat modular protein capable of influencing varied processes in host RBC. This finding is schematically detailed in Figure 3, which exemplifies the participation and the influence of the parasitic processes and pathways on the host cellular roles. The central sliced doughnut in the figure enumerates the parasite proteins under each functional category, and the number of host–-parasite interactions influencing host cellular processes is represented as bars corresponding to each slice of the doughnut. Majority of the host–-parasite interactions, as depicted in the figure, are mediated by parasite proteins participating in erythrocyte rosetting. Indeed, these proteins belong to the hypervariable
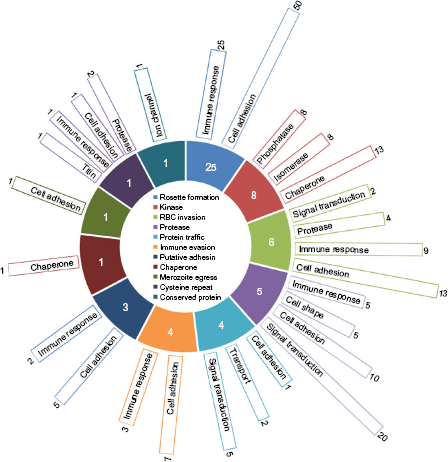
Influence of host-parasite interactions on biological processes and pathways. The central sliced doughnut represents 59
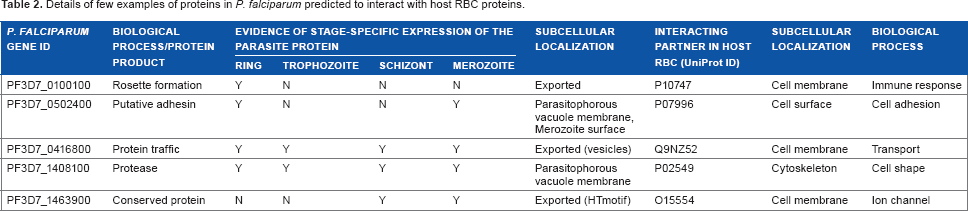
Thus, our structure-based approach has the potential to complement established experimental findings and could provide suitable grounds to warrant an experimental follow-up. Table 2 summarizes the selected examples of interest. The complete list of putative RBC-parasite interactions is provided in Supplementary Table 1. Comparison with previously published computational studies on the identification of host-parasite protein–-protein interactions16,23 yielded a set of five interactions mediated by three parasite proteins and four host proteins, which concurred with our predictions made. Interestingly, we also recognized three host–-parasite interactions that concurred with experimental observations. These include interactions mediated by three erythrocyte-binding antigen proteins of the parasite, which bind to erythrocytes in a sialic acid-dependent manner. 51 The host-parasite interactions in concordance with earlier studies are highlighted in Supplementary Table 1.
Details of few examples of proteins in
Investigations on selected cases at the molecular level are discussed further.
Case study 1: Establishment of host-parasite protein-trafficking machinery
The parasite protein, SAR1 (PF3D7_0416800), is a small GTP-binding protein of 192 amino acid residues, which is involved in the crucial step of budding reaction in vesicle-mediated secretory pathway. Based on our protocol, we recognized one protein from host RBC as plausible interacting partner of SAR1. The predicted interaction between SAR1 and the host ADP-ribosylation factor-binding protein GGA3 (Q9NZ52) is further investigated. The proteins SAR1 and GGA3 were recognized to be evolutionarily related to a protein complex (GTP-bound ADP-ribosylation factor, ARF-GTP, and GAT domain of ADP-ribosylation factor-binding protein GGA1) that demonstrates molecular basis of membrane recruitment of adaptor proteins such as GGA by ARF-GTP. This protein complex elucidated for a mammalian system plays a key role in vesicular transport by docking the adaptor protein GGA1 to membrane for increased efficiency in recognition of cargo receptors. 52 The GAT domain of GGA1 reportedly undergoes conformational change to interact with ARF-GTP. The helix-loop-helix structure, acquired by the disordered N-terminal region of GAT domain, interacts with an interswitch region formed by two antiparallel P strands of ARF-GTP. This ARF-binding disordered region is conserved across the GAT domains of human GGAs, as demonstrated earlier. 52
To assess the molecular and mechanistic details of the host-parasite interaction mediated by the protein pair GGA3-SAR1, the disordered region (166 –-210) of the 723 residue protein GGA3 was modeled using MODELLER v.9.14
53
with the help of template helix-loop-helix structure of GAT domain of GGA1, while reliable structural model for SAR1 (region: 22–-191, model coverage: 89%) was obtained from ModBase, which is a large-scale comprehensive database of comparative protein structure models.
54
The models built were assessed for local structural matches with respect to the template protein complex using TM-align.
55
The program assigns TM-score for a structurally aligned protein pair, which typically acquires a value in (0, 1]. A TM-score of ≥0.50 corresponds to convincing structural similarity, and a TM-score of <0.30 depicts random structural matches.
56
Table 3 provides an account of sequence and structural assessment of the host-parasite protein pair under investigation. As depicted in Table 3, a TM-score >0.9 could be achieved for both host and parasite protein structural models. Thus, GGA3-SAR1 protein-protein interaction was modeled using the template protein complex and a subsequent energy-minimization step was pursued using GROMACS (Version 4.5.5)
57
to achieve a stable form of the modeled complex. The putative host-parasite protein complex was then assessed for the conservation of interfacial residues. Figure 4 highlights the key conserved residues in the predicted GGA3-SAR1 complex. The predominant participation of hydrophobic residues at the interface, as shown in Figure 4, is similar to the hydrophobic interactions observed at the interface of the template protein complex,
52
thus, suggesting usefulness of the predictions made. Additional comparative assessments in terms of shape complementarity and interaction energies at the interfacial region of GGA3-SAR1 protein complex were also pursued to evaluate our predictions further. We employed a shape correlation statistic
Details on sequence and structural assessment of the predicted host-parasite protein pair GGA3-SAR1.


Assessment of putative host-parasite protein pair GGA3-SAR1. (A) Sequence alignment of GAT domains of GGA3 and GGA1 (1J2J:B) and of SAR1 and ARF-GTP (1J2J:A) is described. The conserved interfacial residues are indicated with black arrows. (B) Probable binding pose of the predicted host-parasite interaction is shown in the illustration on the left panel, while the figure in the right panel delineates the residues participating in the GGA3-SAR1 interaction. The structures in Figure 4, 5, and 7 are generated using PyMOL (http://www.pymol.org/). 71
The significance of SAR1 in establishing protein-trafficking machinery in infected erythrocytes has been well demonstrated earlier. It has been postulated that SAR1 gets translocated to erythrocyte cytosol through a specialized secretory organelle, where it participates in trafficking proteins to erythrocyte membrane.60,61 These established observations are in successful accordance with the proposed GGA3-SAR1 protein-protein interaction, thus, implying functional relevance of probable host RBC-assisted protein-trafficking machinery brought about by the parasite.
Case study 2: Strategies acquired by the parasite to proliferate in the host environment
Calcium, a well-studied intracellular messenger in eukaryotes, is known to play a significant role in the regulation of diverse cellular processes and interactions. Several calcium ion-mediated processes are facilitated by the calcium-binding protein calmodulin. One such process is the regulation of cell membrane potential by calcium-activated potassium channels. The proper functioning of potassium channels aids in the regulation of intracellular osmolarity, membrane potential, and electrochemical gradient, thus, forming an integral part of cellular viability. 62 The chemomechanical gating mechanism for such potassium channels has been elucidated at the molecular level, earlier, 63 where calcium-bound calmodulin reportedly binds to the channel and triggers its opening. The crystal structure of calmodulin-potassium ion channel complex elucidates the heterotetrameric association of calmodulin and calmodulin-binding domain of ion channel, that is, dimer of heterodimer. The oligomeric state of calmodulin and calmodulin-binding domain is a dimer in the absence of calcium ions, while the binding of calcium ions to calmodulin triggers the formation of an elongated heterotetrameric complex (Fig. 5), resulting in a rotary movement of the two calmodulin-binding domains, thus, serving as gates to drive open the channel. 63 Based on the structure-influenced approach, we identified probable interaction between host calcium-activated potassium channel protein 4, KCNN4 (UniProt ID: O15554), and conserved parasitic protein of unknown function, PF3D7_1463900.

Crystal structures of calmodulin and calmodulin-binding domain are shown in ribbon representation (PDB code: 1G4Y). The blue ribbon represents calmodulin-binding domain, and the red ribbon represents calmodulin with calcium ions depicted as green spheres.
PF3D7_1463900, a conserved parasitic protein of unknown function of 1071 amino acid residues, was identified to constitute an EF-hand domain region (calcium-binding protein) at its C-terminal end (896-1054). A reliable structural model, using MODELLER, could only be obtained for this region; however, the secondary structural content of the protein was determined to be 76% helical.
64
Likewise, a reliable structural model for calmodulin-binding domain of the 427 amino acid residue protein channel KCNN4 was retrieved from ModBase (region: 304-377). Since, the template protein-protein complex represents the interaction between calcium-binding domain and calmodulin-binding domain, we pursued the analysis on calcium-binding domain or EF-hand domain of the parasite protein and calmodulin-binding domain of host RBC protein channel KCNN4. The binding pose as observed in the template complex could not be directly extrapolated onto the host-parasite protein pairs, owing to the absence of conservation of interface residues. Thus, we probed the probable binding pose of the host-parasite complex with the help of protein-protein docking program ClusPro2.0.
65
ClusPro2.0 identifies a large number of docked conformations, rigorously evaluates energies of each of the docked protein pairs, and recognizes modeled complexes with near-native conformations, which are usually present in the top-ranking clusters.
66
The putative low-energy docked conformation of the host-parasite protein pair, thus obtained, was probed in terms of surface complementarity and interaction energy of the complex. The calculations on geometrical packing at the interface of the predicted RBC-parasite complex yielded an
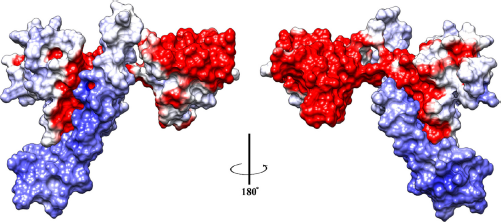
Perspective view of electrostatic complementarity of the modeled host-parasite complex. The host RBC and parasite proteins are rendered as molecular surfaces and are colored based on electrostatic potential (blue: positive, white/gray: neutral, and red: negative). The binding pose of the predicted interaction is illustrated, where the host calmodulin-binding domain in blue is anchored to the C-terminal lobe of EF-hand domain of parasite protein. The two panels show 180° perspective view of the electrostatic contacts between positive and acidic surfaces of host and parasite domains, respectively. This figure is generated using UCSF Chimera 65 (http://www.cgl.ucsf.edu/chimera).

Assessing host-parasite interacting surfaces. The binding pose of host calmodulin-binding domain and parasite EF-hand domain is delineated, where the domains are rendered as transparent ribbons and the putative interacting residues as sticks. Each of the salt bridge forming residues and the residues mediating hydrophobic interactions are highlighted separately. The side chains of the residues highlight hydrogen-bond interactions and hydrophobic interactions.
Conclusion
Understanding the intricacies in the strategies acquired by a pathogen to remodel its host-cell machinery for successful colonization and persistence within the host requires understanding of protein-protein interactions across the host and the pathogen. In addition, the construction of protein interaction network for the pathogen can aid in the comprehension of local and global functional relationships within the pathogen, as described earlier,
21
for the multihost parasite
We have demonstrated the usefulness of structure-based approach integrated with various filters in recognizing 208 physicochemically viable protein-protein interactions across 30 host RBC proteins and 59
Author Contributions
Conceived and designed the experiments: NS, PP, VN. Analyzed the data and contributed to the writing of manuscript: GR. Agreed with manuscript results and conclusions: NS, PP, VN, GR. Made critical revisions and approved the final version: NS. All authors reviewed and approved the final manuscript.
Supplementary Material
Supplementary Table 1
Details on probable interacting proteins across