
Abstract
RNA-binding proteins (RBPs) are at the core of post-transcriptional regulation and thus of gene expression control at the RNA level. One of the principal challenges in the field of gene expression regulation is to understand RBPs mechanism of action. As a result of recent evolution of experimental techniques, it is now possible to obtain the RNA regions recognized by RBPs on a transcriptome-wide scale. In fact, CLIP-seq protocols use the joint action of CLIP, crosslinking immunoprecipitation, and high-throughput sequencing to recover the transcriptome-wide set of interaction regions for a particular protein. Nevertheless, computational methods are necessary to process CLIP-seq experimental data and are a key to advancement in the understanding of gene regulatory mechanisms. Considering the importance of computational methods in this area, we present a review of the current status of computational approaches used and proposed for CLIP-seq data.
Keywords
Introduction
RNA regulation is key to understanding the rules that govern gene expression regulation and epigenetic changes. RNA regulation occurs through a variety of mechanisms such as alternative splicing, alternative transcription initiation, and polyadenylation.1,2 The systemic action of several RNA-binding proteins (RBPs) is one of the principal mechanisms of post-transcriptional gene regulation. Moreover, post-transcriptional regulation has an effect on cell function and dysfunction. 3 In fact, the correct action of each RBP and associated expression level has an impact on important processes for cell function such as cell development. At the same time RBPs' dysfunction or loss of function is associated to diseases like neurological disorders4,5 and cancer.6,7
Although, the RBP role and importance are clear, and thousands of RBPs are present in eukaryotes, the mechanism of action has only been studied precisely for a few RBPs. In order to understand the RBPs' mechanism of action, it is important to identify the RBP binding sites, and from these sites the common motif. However, in humans, motifs are only known for 15% of candidate RBPs, 8 and this percentage is even lower for other organisms. Although, this field is of remarkable importance, it remains almost unexplored.
The first experimental techniques used to determine RBPs binding sites were SELEX, 9 RIP-chip, 10 and CLIP 11 (UV crosslinking and immunoprecipitation). However, these experimental techniques require a significant investment in terms of work, effort, and time. Only recently, genome-wide methods have been adapted to study RBPs' mechanism of action. In particular, CLIP-seq protocols combine the action of CLIP and next-generation sequencing (NGS) to derive a transcriptome-wide set of RBP binding sites. 12 There are different CLIP-seq protocols; each one introduces experimental variations to improve the signal to noise ratio.
Computational methods are relevant for CLIP-seq data processing for the following reasons: CLIP-seq dataset size is significant, it is important to exploit all the information present in the experimental data, and in some cases, it is necessary to integrate other sources of information to process CLIP-seq data. Moreover, a quantitative tool to process entirely CLIP-seq data has not been developed. Instead, tools are designed to face specific CLIP-seq data processing steps. In addition, the number of CLIP-seq datasets is growing (Fig. 1). For all these reasons, computational approaches, designed specifically to deal with CLIP-seq data, are important in this field. In particular, computational methods are a key to process CLIP-seq data, facilitate the analysis, and unveil the RBP-specific roles.

Timeline for computational research on RNA-binding proteins (RBPs). We present three indicators: red, the number of structures reported in the protein data bank; green, the number of publications of computational approaches for RBPs; and blue, the number of CLIP-seq data sets in GEO database.
This review presents the current status in computational methods designed for CLIP-seq data. Our intention is to help the reader find the most adapted tools and to motivate the readers to work on current challenges and necessities. In particular, we provide the reader with the basic background, and we present a brief overview on CLIP-seq experimental protocols and databases that contain CLIP-seq data. Moreover, we present a general computational pipeline to process CLIP-seq data and available methods at each step of the pipeline. Finally, we present future directions and current challenges.
Background
Even though research on RBPs started in the 1970s, the interest in RBPs is concentrated mainly in the last two decades (Supplementary Fig. S1). Reviews on different topics regarding RBPs are present in the literature. We found several reviews regarding the current understanding of RNA regulation 13 17 and the RBPs role in post-transcriptional regulation. 18 20 In addition, interesting reviews are also available on the RBPs role on different organisms 21 23 and comparative studies of RBPs functionality and presence on different organisms.8,24
It is worth noting the reviews on the effect of RBPs on biological processes and diseases. In particular, RBP gain of function and loss of function are associated to important diseases,6,25 like neurological diseases 4 and cancer. 7 The above mentioned reviews highlight the importance of RBP action on gene expression regulation.
Nevertheless, major advances in RBP research are markedby developments of experimental and computational techniques.26,27 In fact, we found several reviews concerning advances in the experimental field.28,29 In Refs. 12 and 30, the authors address the importance of in vivo data and the differences between in vitro and in vivo data. In Refs. 31 and 32, the authors present CLIP, crosslinking, and immunoprecipitation based approaches, and thus, the integration of high-throughput sequencing to in vivo experimental techniques.
On the other hand, reviews on computational approaches for RBP are mainly focused on structure prediction of Protein-RNA interactions,33,34 as structure is key in this type of interactions. 35 39 Nonetheless, two recent reviews bring the attention to the necessity of bioinformatic approaches to process data provided by CLIP-seq protocols.40,41 In, Ref. 40 the authors present bioinformatic approaches designed for transcription factors that are frequently used to process RBP data. 40 Instead in Ref. 41, the authors emphasize the importance of modeling RNA secondary structure to recover RBP, and they conclude that RBP motif recovery is a rapidly expanding field but still in its infancy.
Considering the above, we use three indicators and associated timelines (Fig. 1) to present the focus of computational methods for RBPs. We divided the timeline into two periods, before and after the introduction of CLIP-seq protocols. In the first period, we observe a similarity between the first and second indicators trends. Instead, during the second period, we observe a similarity between the second and third indicators trends. We note that during the second period, computational proposals are essential to handle and process CLIP-seq data. Computational proposals, designed for RBPs, face two main challenges: (1) to provide insights into the RBP-RNA interaction structure and (2) to enable and facilitate CLIP-seq data processing. This review presents the computational methods designed specifically for the second challenge.
Several reviews regarding experimental advances, in particular reviews regarding CLIP-seq protocols, are currently available. On the other hand, reviews regarding computational approaches for RBPs are focused on structural prediction. However, there are no reviews focusing on computational proposals designed for CLIP-seq data, even though this is a rapidly evolving field. For these reasons, we present a review on the current status of computational proposals used and designed for CLIP-seq data. The intention of this review is to present current status, and also to motivate the readers to work on current challenges and necessities.
CLIP-Based Experimental Data
A major step to understand the RBP role is to identify the RBP targets by locating the regions where the protein binds (also known as RNA recognition elements, RRE). The experimental field has achieved notable advances. In particular, experimental techniques used to derive RBP-RNA interactions in vivo are integrated with NGS technologies. As a result, it is possible to derive interaction sites on a large scale. Currently, two experimental approaches, RIP-seq and CLIP-seq, perform such integration.
RIP-seq is the combined action of RNA immunoprecipitation (RIP) 42 and RNA-seq. RIP-seq is used to recover interaction sites between RNA and specific RBPs. 32 Even though RIP-seq is simple, false positives are a major drawback. On the other hand, CLIP-seq approaches combine UV crosslinking and immunoprecipitation with NGS technologies to recover the interaction sites between RBP and RNAs. The use of UV crosslinking makes it possible to obtain reliable sites with a higher level of resolution compared to RIP-seq results.32,41
Considering the focus of this paper, we present CLIP-based approaches in greater detail. CLIP has been widely used to identify the RBP-RNA interaction regions, but this technique alone yields a reduced set of sequences containing the binding regions. 43 In practice, CLIP techniques use UV irradiation to covalent crosslink the RBP-RNA interaction; consequently the investigated protein is immunoprecipitated to isolate the complex, and partial RNase digestion of the bound transcript is used to select a short region of RNA attached to the protein. Nevertheless, only the joint action of CLIP with NGS makes it possible to obtain a transcriptome-wide set of interaction regions. 44 However, there is limited understanding of the crosslinking specificity at a physical level. 1 In order to overcome this limitation, ie, to identify in detail the crosslinking site and to improve the signal to noise ratio, several protocols have been proposed to determine the crosslinking sites: iCLIP, 45 PAR-CLIP 46 and HITS-CLIP 47 49 In particular, the reverse transcription frequently stops at the crosslinking site in the iCLIP protocol. In HITS-CLIP, a nucleotide deletion is frequently found at the exact crosslinked amino-acid. 47 Alternatively, the PAR-CLIP protocol introduces an experimental variation at the beginning of the procedure, in order to facilitate the recognition of the interaction sites. 50 The nascent transcripts are labeled with a photo-reactive nucleoside (4-thiouridine) to print signatures inside and in the vicinity of the crosslinking site. In PAR-CLIP, the thymidine (T) to cytidine (C) transition (if nucleoside used is 4-thiouridine) near the crosslinking site is frequently found.
However, CLIP-seq experimental data need to be processed for further analysis. Therefore, computational methods for CLIP-seq data processing are necessary.
Databases with CLIP-Based Data
We present CLIP-seq data repositories that are publicly and freely available. This information is helpful to check whether there is another CLIP-seq dataset for the same protein under study or protein family. In addition, this information is quite valuable to design, test and validate new computational proposals.
Data sets obtained with CLIP-based protocols are frequently uploaded to public databases such as the Gene Expression Omnibus (GEO from NCBI) 54 and ArrayExpress (from EBI). 51 The authors of CLIP-based studies upload experimental data sets to share obtained results. In particular, the uploaded data often contain raw and processed data and a brief description of the data set. GEO and ArrayExpress are international public repositories that store high-throughput data obtained by the research community; the data sets uploaded in these two databases are publicly and freely available.
Recently, three databases have been developed specifically to store CLIP-based data: CLIPZ, 52 doRiNA, 53 and starbase v2.0. 55 These databases store CLIP-seq data, either uploaded directly by researchers or data sets available in the repositories previously mentioned. The data sets stored are carefully revised and processed. In addition, these databases provide additional useful functionalities for researchers in the field. In Table 1, we present the principal characteristics associated with the three databases. In particular, we present the data stored, the total number of data sets, and a graph with the organisms present and the protocols used. Moreover, we provide a summary of the main advantages associated to each database, the website link, and reference.
Databases with CLIP-seq data and associated characteristics.
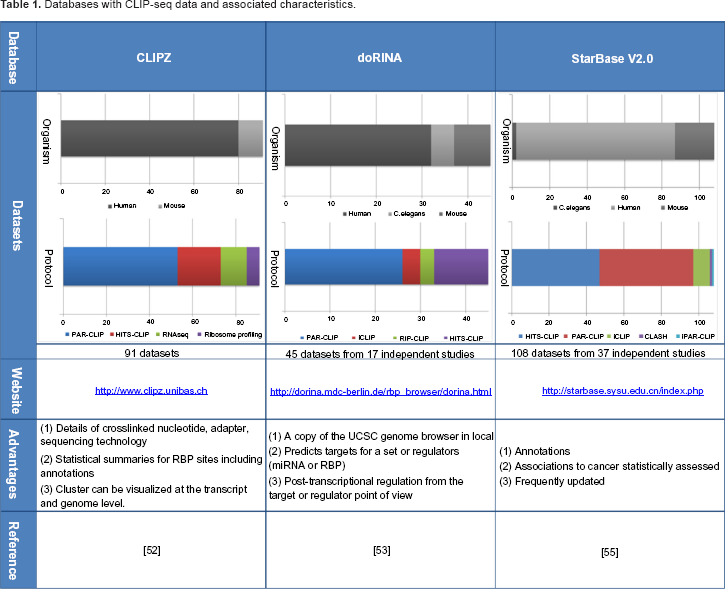
CLIPZ is the first database published, specifically designed for CLIP-based data. 52 The database contains data sets published from 2010 to 2013. Most of the data available are CLIP-seq data, however, the database contains RNA-seq data from the same samples and Ribo-some profiling data. The CLIPZ database provides details for further processing such as the crosslinked nucleotide, the adapter, and the sequencing technology. In addition, the database provides statistical summaries, such as region preference, annotation summary, mutation plots, read quality, and read clusters length. The statistical summaries are presented for each data set or simultaneously for several selected data sets, which is particularly useful to make comparisons among several datasets.
doRiNA 53 is the second database published. This database contains data from 2010 to 2012, mainly CLIP-based data, but it also contains a few RIP-CHIP data sets. doRiNA has a local copy of the UCSC genome browser, which makes possible to have access to UCSC tracks. It is worth noting that doRiNA gives a post-transcriptional regulation view from the target or the regulator (miRNAs or RBP) point of view.
The third database is starbase v2.0. 55 This database contains data from 2010 to 2013. Initially, starbase was designed for microRNAs but the new version contains CLIP-seq data for a variety of RBPs. The database provides annotations for RBP sites, in particular lnc-RNA, mRNA, pseudogenes, and sncRNA. Moreover, it shows RBPs possible associations to cancer, which are statistically significant.
These freely available databases provide access to CLIP-based datasets. However, it is necessary to process the available data.
Computational Pipeline to Process CLIP-Seq Data
Even though there is great room for further improvements in CLIP-seq data computational processing, several bioinformatic approaches have been proposed so far. The current proposals address several steps in CLIP-seq data processing. In Figure 2, we present a pipeline with the most important steps in CLIP-seq data processing, and we associate computational tools designed for each step.

Steps for CLIP-seq data processing.
Here, we summarize these computational approaches for processing CLIP-seq data. We divided the computational approaches into categories depending on the scope. Moreover, in Table 2, we present additional characteristics for computational approaches specifically designed for CLIP-seq data.
Computational proposals specifically designed for CLIP-seq data processing.

Read Mapping and Cluster Detection
The first step in CLIP-seq data processing is to map all the reads to the genome and transcriptome. During this step, at least one mismatch should be allowed because the experimental protocols induce nucleotide transitions (also known as mutations). Usually, the most frequently used algorithms to perform this step are Bowtie, 56 RMAP, 57 and Novoalign. 58 However, TopHat59,60 is commonly used at this step, to identify exon-exon junctions.
Once the sequence reads are aligned to the genome and transcriptome, the following step is cluster detection. A cluster of reads is a group of reads, where a read belongs to a cluster if it overlaps at least one nucleotide to another read from the cluster. Several restrictions can be used to filter noise at this step. Usually, only reads with a length higher than a determined threshold are considered. In addition, clusters with a minimum number of unique reads are selected for binding site detection.
Binding Site Detection
After the cluster detection, the following step is reliable binding site detection. The main challenge at this step is to improve the signal to noise ratio, hence to remove background and false positives. The most common strategy to face this challenge is to analyze clusters distribution profiles. Computational approaches that use this strategy are WavClusteR, 61 PARalyzer, 62 Piranha, 63 PIPE-CLIP, 64 and dCLIP 65 However, considering RNA structural features is also a good strategy that is present in GraphProt. 66 In this section, we briefly describe the above-mentioned computational proposals and present specific advantages.
It is worth noting that at this step it is definitely a plus to consider the number of sequences aligned to a specific cluster because this number strongly depends on the transcript abundance and cluster length.63,65
PARalyzer 62 is the first computational approach designed for RBP site detection. This tool uses a non-parametric kernel density estimate and a classifier; it identifies the RBP sites based on a combination of T to C mutations and read density. PARalyzer can improve binding site recognition in data sets published.
WavClusteR 61 is a computational tool proposed to overcome two problems in PAR-CLIP data processing. The first problem is the number of false positives, and the second is to improve cluster detection. Mutations present in experimental data are experimentally induced and also non-experimentally induced. In fact, nucleotide mutations are induced by the experimental protocol but in addition, several other factors cause mutations as well, such as sequencing errors, contamination with external RNA, and single-nucleotide polymorphisms (SNPs). WavClusteR uses a non-parametric two-component mixture model to distinguish experimentally from non-experimentally induced mutations, thus reducing the presence of false positives. In addition, the second part of WavClusteR exploits geometric properties of the coverage function to identify reliable binding sites.
Piranha 63 is a computational tool designed for site identification (peak calling), in CLIP-seq (HITS-CLIP, PAR-CLIP, iCLIP) and RIP-seq data. Piranha deals with three key challenges on computational site identification: (1) presence of noise and false positives, (2) resultant reads depend on transcript abundance, and (3) it is important to integrate different sources of information to improve peak calling. Piranha 63 uses a zero-truncated negative binomial distribution to model read counts, when additional information is available (covariates such as the transcript abundance), Piranha uses a zero-truncated negative binomial regression model. In addition, Piranha can compare CLIP-seq data from different samples because it corrects the reads dependence on transcript abundance.
PIPE-CLIP 64 is a pipeline to identify binding regions. In PIPE-CLIP, the data are pre-processed to remove noise such as the PCR duplicates. Consequently, PIPE-CLIP identifies enriched clusters (considering cluster length effect on the number of reads) and reliable mutations. Each enriched cluster with at least one reliable mutation is selected as an RBP binding site.
dCLIP 65 is a computational approach designed for quantitative CLIP-seq comparative analysis. dCLIP has two parts: normalization and RBP sites detection for comparison. The normalization step is necessary for an unbiased comparison. The second part is necessary to detect common or different sites for different CLIP-seq samples in order to perform a comparison.
GraphProt 66 is a machine learning approach designed to identify RBP binding sites. This approach uses a training set to learn RBP binding preferences from high-throughput experimental data such as CLIP-seq and RNAcompete. 67 It uses a graph-kernel strategy to obtain a large set of features from the training set and any input data set. It should be noted that the features concern RNA sequence and also structure characteristics. GraphProt uses a support vector machine (SVM) to identify RBP sites using the set of features extracted. Moreover, when affinity data are available, GraphProt uses a support vector regression (SVR) to estimate affinities.
Motif Recovery
The next step is to search the specific motif recognized by the RBP, among reliable binding sites. So far, two strategies are used for this purpose. The first one consists in using tools developed to detect motifs in DNA that consider only sequence information. The most frequently used tools for this strategy are MEME, 68 cERMIT, 69 GLAM2, 70 and MatrixREDUCE. 71 The second strategy consists in using motif recognition algorithms that integrate additional information to guide the motif search. Examples from the second strategy are MEMEris,72,73 PhyloGibbs,74,75 RNAcontext,76,77 and RNAmotifs. 78
It is worth noting that the second strategy permits to consider RNA-specific characteristics. In fact, MEMEris 72 uses RNA secondary structure to guide the motif search toward single-stranded regions, and PhyloGibbs 74 integrates conservation information. Moreover, RNAcontext 77 works on large-scale RNA-binding affinity datasets and provides the RNA motif in terms of sequence and structure. Finally, RNAmotifs 78 identifies multivalent regulatory motifs.
Global Prediction
Once the RBP has a defined motif, we can use the motif to predict binding sites in a determined species. For this purpose, we should analyze motif occurrence characteristics 79 and predict candidate RBP binding sites. mCarts 80 and GraphProt 66 are two approaches proposed for this purpose.
In particular, mCarts is an algorithm based on a hidden Markov model that predicts functional RBP binding sites based on the number and spacing of motif sites, accessibility (RNA secondary structure) and conservation information. On the other hand, GraphProt is a machine learning approach that predicts candidate RBP binding sites within the same organism (training set data). In Table 2, we present additional characteristics such as availability.
Considerations to Reduce False Positives
In this section, we present two studies on CLIP-seq data, the results can be added to computational tools to reduce the false positives and improve the performance.
As already mentioned, in CLIP-based protocols covalent bonds are induced through UV crosslinking, only the RNA sites with strong bonds are selected through stringent washes. A study on PAR-CLIP background is presented in Ref. 81. This study presents possible sources of false positives such as RNAs bound to proteins different from the RBP of interest or false crosslinking events. Moreover, it shows that quantifying and taking into consideration possible sources of false positives are important to improve the recognition of the site specificity. As a result of the study, a set of background binding events in PAR-CLIP data is publicly available in GEO (GSE50989).
In addition, CapR is a tool designed to obtain a structural profile, which has been applied to CLIP-seq data. CapR 82 obtains a probability for each RNA base position, which reflects the location at determined structural contexts (CapR defines six contexts). Using these probabilities is possible to obtain a structural profile for an RNA sequence. Researchers can apply CapR on CLIP-seq data, so far, the obtained results are encouraging.
Further Considerations
As indicated above, computational approaches are key to process CLIP-seq data and this field is expanding. In the RBP-RNA action, not only the sequence is important but also the RNA secondary structure. RBPs recognize the motif sequence content as well as the motif secondary structure. Even though RNA secondary structure is considered in a few tools, it is a must and not just a plus.
Moreover, additional improvements can be achieved by integrating information about RBP domains, such as the one present in RBPDB database. 83
In addition, advances on experimental field have provided techniques such as RNAcompete. 67 RNAcompete provides an affinity measure, independent on transcript abundance, it is an in vitro method. However, there is not a computational proposal that integrates information from both CLIP-seq and RNAcompete data, simultaneously.
Finally, an additional step can be added to the pipeline in Figure 2. After global prediction, integrative approaches for network inference can be used. This step is necessary to have a complete understanding of the specific role of RBPs.
Author Contributions
PHRH conceived the idea and wrote the manuscript. PHRH and EF jointly developed the structure and arguments for the paper. EF made critical revisions. Both authors reviewed and approved of the final manuscript.
Supplementary File
Supplementary Figure S1
Timeline for research on RNA binding proteins, this figure shows the number of publications on RNA binding proteins reported on PubMed since 1972.