
Abstract
Matrix-Assisted Laser Desorption/Ionization time-of-flight (MALDI-TOF) mass spectrometry is evaluated as an elucidation tool for structural features and molecular weights estimation of some extracted herbaceous lignins. Optimization of analysis conditions, using a typical organic matrix, namely α-cyano-4-hydroxycinnamic acid (CHCA), in combination with α-cyclodextrin, allows efficient ionization of poorly soluble lignin materials and suppression of matrix-related ions background. Analysis of low-mass fragments ions (
Keywords
Introduction
Lignin is the cell wall-resistant component of vascular plants and represents 15% to 30%, by weight, of the lignocellulosic feedstock. 1 Lignin is therefore the third most abundant biopolymer on earth after cellulose and hemicellulose. In the context of an integrated biorefinery concept, it is important to upgrade the entire lignocellulosic material. 2 Whilst the production of fermentable sugars, platforms and building blocks from both cellulose and hemicelluloses is extensively developed, the non-energetic valorization of lignin remains a major challenging task. 3 The final application of lignin can indeed significantly vary as a function of both extraction methods and type of lignocellulosic sources. The development of fast, non-invasive, and reliable methods for the physicochemical characterization of extracted lignin is thus a prerequisite to optimizing processing conditions such that they are adequate.
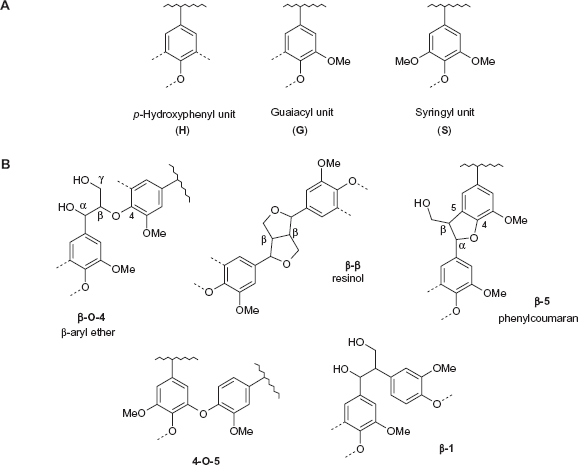
Native lignin is an amorphous, three-dimensional cross-linked heteropolymer consisting of phenylpropanoid units joined together by several specific ether or carbon-carbon bonds.
4
Lignin is composed of three phenylpropane derivatives in varying ratios, referred to as p-hydroxyphenyl (H), guaiacyl (G) and syringyl (S), linked by β-O-4, β-β, β-5 (phenylcoumaran), 4-O-5 or β-1 bonds in various bonding patterns (Fig. 1). Practically, lignin can be extracted from lignocellulosic feedstocks by physical and/or chemical processes that modify its native structure. Ether linkage of the β-O-4 type is the most frequent in herbaceous plants and its solvolysis under formic acid/acetic acid (Delmas) conditions is used to depolymerize lignin, as carbon-carbon bonds are more resistant to degradation. Conversely, under alkali conditions, ie, ammonia soaking, phenylcoumaran substructures are easily broken whilst β-O-4 linkages remain almost unaltered.
5
(
Because value-added applications of extracted (depolymerized) lignins are envisioned, complete structural characterization of lignins is required to ensure a better comprehension and prediction of structure-functions. Due to lignin structural heterogeneity, traditional spectroscopic methods still reveal a fascinating complexity. Liquid nuclear magnetic resonance (NMR) spectroscopy, including two-dimensional techniques and quantitative 13CNMR, provides extensive information of the salient structural features, mainly monolignols (G/S/H) and/or end-groups distribution, and quantification of inter-unit linkages.6–8 Although strikingly useful, the obtainable signal-to-noise ratio for these analyses is only improved for high (derivatized) lignin concentrations (up to 50 mg/mL) in analysis solvents combined with long acquisition times (up to several days). The propensity of lignins to associate and aggregate in organic solutions, identifying apparent molecular weight changes over time and possible sedimentation of the samples during analysis, is another drawback of wet analysis methods. This aggregation behavior has been evidenced using static light scattering photometric (MALLS) measurements, and is notably dependent on the nature of the analysis solvent.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) is commonly used for the characterization of synthetic or natural polymers.
9
Information such as average molecular weights (weight and number;
Due to their structural composition, their formal charge and/or their molecular weights, lignins are sometimes poorly soluble in classical analysis solvents (for example, water, chloroform, and toluene). The quest for a fast technical method for their investigation, without prior chemical derivatization that might deteriorate lignin structure, has prompted us to investigate MALDI-TOF-MS as a potential solution. This mass spectrometry methodology is non-destructive and, conversely to traditional wet chemical analytical tools, can possibly reflect the entire lignin sample composition. The scope of this work is to describe a versatile and straightforward analytical protocol for lignin mass fingerprint analysis and average molecular weight estimation. Miscanthus (
Experiment
Lignin materials
Several herbaceous isolated lignins were selected for this study. These technical lignins were characterized by a S/G ratio of about 1/1 as estimated by quantitative 13C NMR.
5
Three selected lignin fractions (FAL107, FAL80 and AL) were extracted from
The solubility of lignin samples was determined gravimetrically by filtration (0.45 μm filters) of the suspension (10 mg/mL lignins in organic solvent or water) after 1 hour of stirring at room temperature. Solubility of unmodified FAL107 was inferior to 0.1 g/L in water (18 °C, pH 6). In non-polar solvents such as toluene or chloroform, its solubility was measured at about 3.2 and 1.5 g/L respectively. In polar aprotic solvents such as acetone, its solubility was slightly improved with a value of 5.6 g/L. For FAL80 and AL, this solubility in acetone was of 6.4 and 1.2 g/L respectively and of 3.9 and 0.8 g/L for SG-107 and SG-AL.
Molecular weight determination using HPSEC-RI
Lignin molecular weights were estimated using size-exclusion chromatography (SEC) and were expressed as polystyrene-equivalent molecular weights (Mp). 14 Three Styragel columns (HR1, HR2 and HR3; Waters) were connected in series to an HPLC system (Agilent Technologies, 1200 series) equipped with a differential refractometer (BI-DNDC/GPC, 620 nm) using THF as the eluent (flow rate, 1 mL/min). Analyses were performed in duplicate. Calibration was ensured using commercial polystyrene standards. Measurements were performed on acetylated or non-acetylated lignins dissolved in THF (1 mg/mL). Acetylation was achieved according a published protocol. 5
MALDI-TOF-MS instrumentation and conditions
High resolution mass spectra were recorded on an Ultraflex MALDI time-of-flight mass spectrometer (Bruker Daltonic) operating in the reflector and linear positive or negative mode using a pulsed nitrogen laser (λ 337 nm, pulse rate of 10 Hz) as the ionization source. The analyzer was used at an acceleration voltage of ±20 kV Laser light was focused on the sample using a 5.2 kV lens. A pulsed ion extraction was optimized to 200 ns. A peptide calibration set was used as the external standard. For each lignin sample, MALDI-TOF-MS was ensured on 10 distinct sample deposit zones. A total of 300 shots were provided. Each sample preparation and analysis were duplicated.
α-CD/CHCA matrix preparation
α-CD/CHCA matrix was prepared by mixing 240 mL of a solution of 10 mM α-CD in water with 40 mL of CHCA 75 mM in acetonitrile/H2O (7/3 v/v) containing 0.1 vol.% trifluoroacetic acid. The complex α-CD/CHCA was obtained after sonication at 50 °C for 1 hour. Interaction between α-CD and CHCA was confirmed by UV-Visible spectroscopy.
Results and Discussion
Optimization of MALDI-TOF-MS for extracted lignins
FAL107 lignin was arbitrarily selected for the optimization process. FAL107 absorbed conveniently at λ 337 nm and could be analyzed without a matrix, as proposed by Yoshioka et al, 15 using a nanostructured silicon-based target. However, in our study, preparation of the lignin sample with a traditional matrix was preferred, as partial oxidation of unsaturation sites was reported under matrix-free conditions and might cause confusions with the presence of some structural features characteristic of lignins. 16
After a screening of conventional commercial MALDI matrices, 17 only aromatic acidic compounds, such as α-cyano-4-hydroxycinnamic acid (CHCA) or 2,5-dihydroxybenzoic acid (2,5-DHB), were found convenient for sufficient lignin-related ions production and adequate mass resolution with our incident λ 337 nm laser equipment ionization source. Best results were also obtained when MALDI-TOF-MS sample preparation was achieved using the “sandwich method”, where the lignin analyte was deposited between two fast evaporated matrix layers. Practically, the lignin is not premixed with the matrix. A lignin sample droplet is applied on top of a fast-evaporated matrix layer (acetone), followed by the deposition of a second layer of matrix in non-volatile solvent (acetonitrile). The performance of CHCA and 2,5-DHB was then examined using four “classical” solvent systems: 30% aq. acetonitrile with 0.1% trifluoroacetic acid (TFA); 10% aq. acetone; 30% aq. acetone; and water-methanol (2:1). Saturated solutions of both matrices were used. Adjunction of sodium chloride was required to ensure suitable sample ionization for FAL107/DHB and addition of TFA was necessary for FAL107/CHCA. MALDI-TOF-MS spectra were acquired in the reflector positive ion mode by randomly irradiating 300 times the sample spot. The pulse ion extraction delay was optimized at 200 ns. Because of its solubility in polar aprotic solvents, FAL107 was prepared in acetone. A concentration of 0.5 to 5.6 mg/mL provided convenient MALDI-TO-MS sensitivity.
Figure 2 shows the best quality spectra obtained for FAL107 using either 2,5-DHB in acetone-water (7:3) in combination with NaCl (Fig. 2A) or CHCA in 30% aq. acetonitrile with 0.1% TFA (Fig. 2B). The MALDI-TOF-MS spectrum for FAL107 sandwiched between two layers of CHCA revealed an oligomeric distribution from about 100 to 600 Da, with a gradual decrease in intensity. Matrix clusters signals appeared in this low mass range and probably interfered with lignin fingerprints. This drawback was also noticed using DHB, and was amplified with the addition of sodium chloride. Taking into account that constitutive monolignols in FAL107 were syringyl and coniferyl alcohols in a 1/1 ratio (average molar mass: 195.2 g · mol-1), the three main oligomeric distributions denoted in Figure 2B were attributed to monomers (denoted [M]), dimers ([2M]) and trimers ([3M]). Assignment of mass signals of these low-mass distributions was partially hampered by inherent matrix-related ions below Reflector positive ion mode MALDI-TOF-MS spectrum of FAL107 with DHB (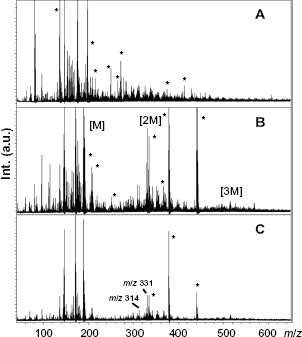
To extend the range of applicability of MALDI-TOF-MS for lignin analysis, we checked a universal lignin/matrix sample preparation strategy based on the encapsulation of CHCA in a cyclodextrin cavity.
18
Then, only the protonated matrix-related ions were detected (mainly at Reflector positive ion mode MALDI-TOF-MS spectrum of CHCA alone (without lignin) (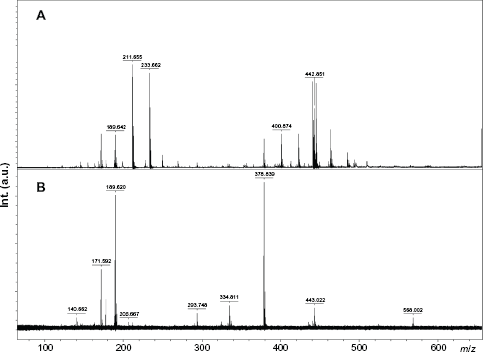
MALDI and HPSEC analysis of technical lignins
HPSEC analysis of FAL107 in THF revealed a mono-modal distribution curve with an apparent molecular weight (Mp) of about 2000 for non-acetylated FAL107 and of 2800 for its acetylated counterpart. MALDI-TOF-MS analysis for FAL107/α-CD/CHCA revealed, however, lower molecular weight compounds ranging from 100 to 600 Da, clearly inferior to the molecular weight measured by HPSEC (Fig. 2C). A depolymerization of the sample under laser beam could be suggested. We decided to explore if MALDI-TOF spectral area between
Constitutive monolignols ions were observed below 200 Da and were detected in the positive ion mode spectrum as their proton and alkali adduct ions. Specific MS peaks were recorded, with variable intensity, at
With Chemical structure of lignin dimeric fragment as proposed by Delmas and Banoub.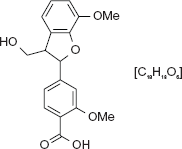
Transfer of this MALDI-TOF-MS extrapolative approach was envisaged for Miscanthus FAL80 and AL lignins (Fig. 5). HPSEC-RI analyses revealed mono-modal mass distributions with Mp values of 1700 for non-acetylated FAL80 and of 2300 for its acetylated analogue. Acetylation of AL was mandatory for HPSEC analysis. An average molecular mass Mp of 3100 was measured. For FAL80 using the MALDI-TOF-MS positive ionization mode, peaks distribution in the Reflector-positive ion mode MALDI-TOF spectra in the 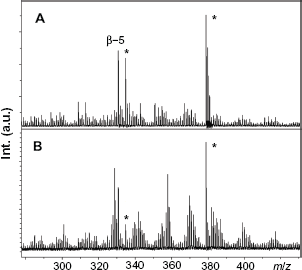
Conversely, MS signal for ammonia lignin AL was exploitable up to about
Our MALDI-TOF-MS protocol with α-CD/CHCA was then sampled for the study of other herbaceous lignins, namely Switchgrass lignins extracted under acidic (SG-107) or alkali (SG-AL) conditions (Fig. 6). SG-107 was characterized by an Mp value of 1600. MALDI MS spectrum in the positive ion mode, using α-CD/CHCA matrix system, revealed a peak distribution ranging from Positive-ion mode MALDI-TOF-MS spectrum of Switchgrass lignin recovered after ammonia soaking (SG-AL).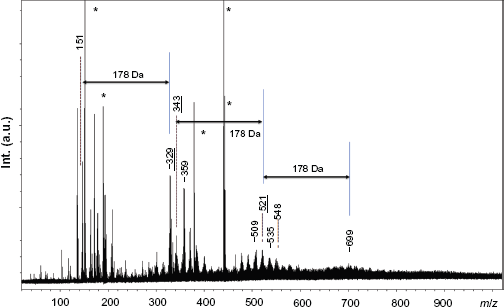
For SG-AL, the best quality spectrum was obtained in the positive ion mode using CHCA alone, instead of α-CD/CHCA, and with an extraction delay of 20 ns. A suspension of SG-AL in acetone, dried between two layers of CHCA, offered convenient results. While the mass range covered by MALDI-TOF-MS was detected between 100 and 1000 Da, the Mp value for THF-soluble SG-AL lignin was measured at 500 Da using HPSEC-RI, evidencing good agreement between both techniques. The mass spectrum of SG-AL, disturbed by the presence of matrix-related peaks, showed a fine oligomeric cluster distribution ranging from monomers (about 180 Da) to tetramers (about 750 Da). In this sample, the hyperfine structure of the spectrum highlighted peaks separated by Δm = 12-14 mass units (ie,
Conclusions
Our results demonstrated that MALDI-TOF-MS was able to provide useful information on the oligomeric distribution of different types of herbaceous lignins containing mainly guaiacyl and syringyl moieties. Involvement of α-cyano-4-hydroxycinnamic acid/α-cyclodextrin as the matrix system allowed efficient sample ionization and suppression of matrix-related signals.
This technique could discriminate between samples extracted using ammonia-soaking and formic/acetic acid conditions with the occurrence of specific “fingerprints”, being mass peaks in the 100-800 Da region. Ionization in the positive ion mode was found to be very efficient for low molecular weight samples, and conveniently highlighted β-5 bonds between phenylpropane units, notably at
Author Contributions
Conceived and designed the experiments: AR, CV, MS, BW, MP. Analyzed the data: AR, CV, MS, BW, MP. Wrote the first draft of the manuscript: AR, CV, MS, BW, MP. Contributed to the writing of the manuscript: AR, CV, MS, BW, MP.
Agree with manuscript results and conclusions: AR, CV, MS, BW, MP. Jointly developed the structure and arguments for the paper: AR, CV, MS, BW, MP. Made critical revisions and approved final version: AR, CV, MS, BW, MP. All authors reviewed and approved of the final manuscript.
Funding
This work was financially supported by the Walloon Region (TECHNOSE Excellence Research Program, project number 716757). The authors also thank the “Fonds pour la Formation à la Recherche dans l'Industrie et dans l'Agriculture” (FRIA) for a fellowship to M. Simon.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Footnotes
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.