
Abstract
Pulmonary arterial hypertension (PAH) is a condition of unknown etiology whose pathological features include increased vascular resistance, perivascular inflammatory cell infiltration and pulmonary arteriolar remodeling. Although risk factors for PAH are poorly defined, recent studies indicate that obesity may be an important risk factor for this condition. The mechanisms leading to this association are largely unknown, but bioactive mediators secreted from adipose tissue have been implicated in this process. One of the most important mediators released from adipose tissue is the adipokine adiponectin. Adiponectin is highly abundant in the circulation of lean healthy individuals, and possesses well-described metabolic and antiinflammatory actions. Levels of adiponectin decrease with increasing body mass, and low levels are directly linked to the development of PAH in mice. Moreover, overexpression of adiponectin has been shown to protect mice from developing PAH in response to inflammation and hypoxia. Based on the findings from these studies, it is suggested that the effects of adiponectin are mediated, in part, through its antiinflammatory and antiproliferative properties. In this review, we discuss the emerging evidence demonstrating a role for adiponectin in lung vascular homeostasis and discuss how deficiency in this adipocyte-derived hormone might explain the recent association between obesity and PAH.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a severe medical condition associated with a sustained elevation of pulmonary arterial pressure. PAH occurs as either a sporadic disease without identifiable risk factors or develops in association with other preexisting medical conditions such as connective tissue diseases, chronic infections (e.g., human immunodeficiency virus) and cirrhosis.[1] To date, the pathogenesis of PAH remains poorly understood, but it is generally accepted that imbalances in vasodilator and vasoconstrictor substances and altered immune, growth and proliferative processes contribute significantly[1]
There are surprisingly few risk factors known to be associated with the development of PAH, but recent epidemiological data suggest that increased body mass index influences the development of this condition.?[2] While the mechanisms linking obesity to PAH are not well established, there is emerging data to suggest a pathogenic role for the adipocyte-derived hormone adiponectin in this process.[3–7] This review highlights the recent data related to adiponectin in lung vascular homeostasis and discusses the potential mechanisms by which hypoadiponectinemia might influence the development of PAH.
Obesity as a risk factor for vascular disease
Obesity is a major public health problem, not least because of its association with increased mortality. The major cause of increased mortality in obese subjects is cardiovascular disease, as obese individuals are at greater risk for developing hypertension, atherosclerotic heart disease and stroke.[8–9] Despite the well-recognized association between obesity and systemic vascular diseases, evidence has only recently emerged for a similar relationship between obesity and diseases of the pulmonary circulation. These data suggest that increased body mass index is linked to the development of both acute and chronic pulmonary vascular diseases. For example, clinical studies have identified increased body mass index as a risk factor for the development of acute lung injury; a condition that results, in a large part, from a loss of endothelial cell barrier function.?[10] In addition, several lines of evidence now suggest that obesity plays a pathogenic role in the development of PAH. Specifically, autopsy studies indicate a higher prevalence of hypertensive changes in blood vessels of the pulmonary circulation of obese subjects when compared with nonobese historical controls.[11] Moreover, findings from the REVEAL registry, the largest pulmonary hypertension database in the United States, indicate a higher prevalence of overweight and obese individuals among those with idiopathic forms of PAH.[2] Notably, this association appears independent of conditions associated with the development of PAH (e.g., diastolic dysfunction, obstructive sleep apnea). Together, these findings suggest that obesity is a condition that globally disrupts vascular homeostasis and predisposes to the development of systemic and pulmonary vascular diseases.
Obesity and adipokines
Over the last several decades, there have been many important discoveries regarding the mechanisms that mediate the health-related consequences of obesity. One key finding has been the observation that obesity is a chronic inflammatory condition, and that persistent low-grade inflammation contributes significantly to the pathogenesis of obesity-related diseases. Adipose tissue is now recognized to be an important endocrine organ through its release of bioactive mediators, called adipokines, and chronic low-grade inflammation develops from obesity-driven imbalances in the secretion of pro-and antiinflammatory adipokines. In lean organisms, these molecules regulate biological processes important to energy homeostasis, inflammation and tissue remodeling. However, excess accumulation of body fat, as occurs in obesity, is associated with adipocyte dysfunction and the altered secretion of these hormones, which in turn contributes directly or indirectly to the development of obesity-related diseases.
Adiponectin is a multi-functional adipokine
Adiponectin is arguably the most important adipokine secreted from adipose tissue because of its pleiotropic actions in metabolism, immune regulation and vascular homeostasis. As suggested by its name, adiponectin is produced almost exclusively by adipocytes and is secreted into the plasma at a high concentration, where it is present at 3-30 μg/mL and accounts for up to 0.01% of the total plasma protein.[12] Circulating forms of adiponectin exist as high molecular weight oligomers, whose molecular weight can exceed 300 kDa, as well as hexamer and trimer structures.[13] The abundance of adiponectin and its ability to form several oligomeric fractions provide a possible explanation for why adiponectin has multiple functions.
Circulating adiponectin levels are decreased in obesity,[12] type 2 diabetes,[14] metabolic syndrome[15] and a variety of cardiovascular diseases.[16–20] Conversely, plasma adiponectin levels are elevated by weight loss,[14] treatment with thiazolidinediones[21] and dietary fish oils.[22] At a cellular level, adiponectin production by adipocytes is impaired by oxidative and endoplasmic reticulum stress and activation by inflammatory cytokines that are prevalent in the adipose tissue of the obese.[23] However, it should be noted that adiponectin levels are elevated, rather than decreased, in a number of chronic inflammatory and autoimmune diseases.[24] The reason for this paradoxical behavior is unknown, but it could be the result of the compensatory regulatory mechanisms or development of an adiponectin-resistant state.[25]
Relevant to this review, adiponectin functions to protect against the development of both metabolic and vascular diseases. The most convincing studies employ mouse genetic models that lack a functional adiponectin gene or transgenically overexpress adiponectin acutely or chronically. In addition, some studies have employed recombinant adiponectin protein preparations, but these experiments are confounded by variations in the quality and structural features of the adiponectin protein preparation obtained from a different source.[26] Overall, studies suggest that when compared with wild-type mice, adiponectin-deficient mice are prone to metabolic and vascular diseases. For example, adiponectin-deficient mice develop a more severe insulin-resistant state when fed a high-calorie diet.[27,28] On the other hand, transgenic overexpression of adiponectin in obese ob/ob mice leads to a normalization of glucose despite a massive increase in fat depot size.[29] With regard to vascular disease, the majority of studies have focused on the systemic circulation. In these studies, adiponectin has been found to be protective in models of myocardial ischemia-reperfusion injury,[30] pressure overload cardiac hypertrophy,[31] cardiac remodeling associated with heart failure,[32,33] hypertension,[34] peripheral artery disease[35,36] and stroke.[37]
At a cellular and molecular level, adiponectin appears to exert these effects by acting on a variety of cell types (Fig. 1). For example, adiponectin will confer an antiinflammatory phenotype in macrophages[38] and vascular endothelial cells,[39] and stimulate catabolic pathways in liver and muscle.[40,41] These actions are mediated, in part, through the ability of adiponectin to activate intracellular signaling pathways via its interaction with the cell surface receptors AdipoR1 and AdipoR2.[42] However, as noted previously, adiponectin circulates at unusually high levels and it undergoes multimerization into high molecular weight complexes. These features suggest an atypical ligand–receptor interaction. In this regard, it has been recognized that adiponectin will bind to the GPI-anchored cell surface protein T-cadherin that localizes adiponectin to the vascular endothelium.[43] Denzel et al. recently showed in murine genetic models that T-cadherin is essential for adiponectin-mediated cardioprotection.[44] In addition, the structural similarity of adiponectin with the collectin family of proteins suggests that it may be functionally similar to this class of proteins as well. Consistent with this hypothesis, it has been shown that adiponectin can suppress inflammation by facilitating the uptake of apoptotic cell debris by macrophages, a property that is shared by other collectin proteins.[45]
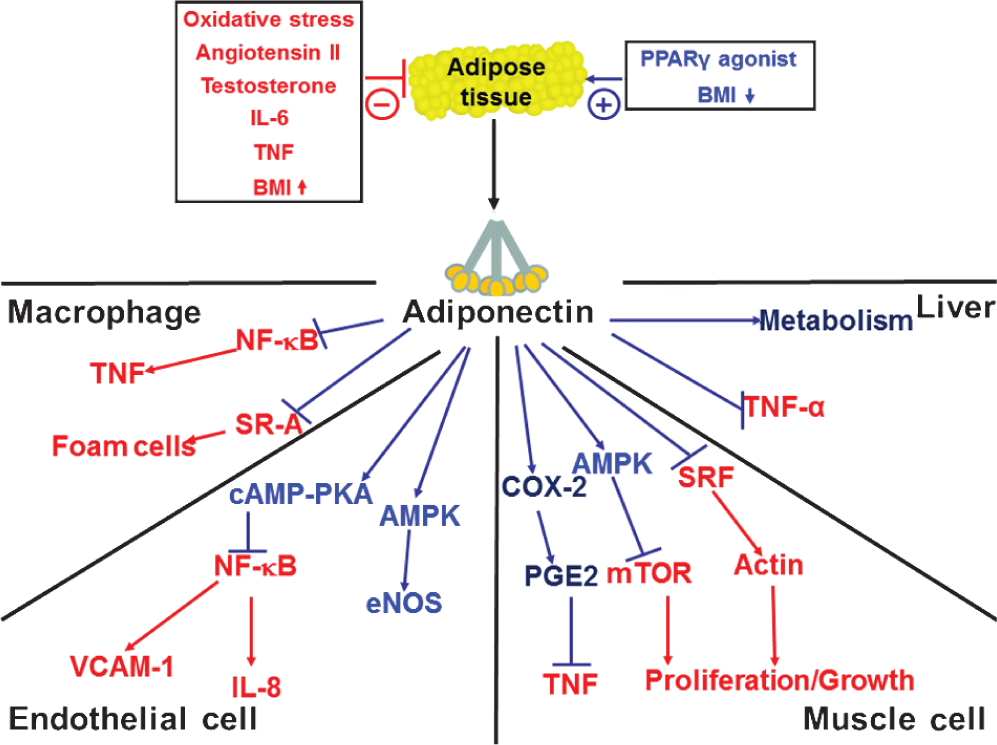
Effects of adiponectin on key cellular targets.
Pathophysiological mechanisms of pulmonary arterial hypertension
The pathogenic mechanisms that lead to PAH are complex, but ultimately result in functional and structural changes to the pulmonary vasculature. Notably, in most forms of the disease, there is accumulation of immune and vascular cells (endothelial cells and pulmonary artery smooth muscle cells) within the arterial lumen, and this is associated with vascular remodeling and changes in vascular tone.[46] How adiponectin potentially modifies these processes is discussed in the following sections.
Adiponectin is a modulator of vascular tone
Classically, PAH was thought to develop from an imbalance in vasodilator and vasoconstrictor substances. Although this is no longer thought to be the only pathogenic mechanism of PAH, increased vascular tone is often an important feature of this condition. In addition, most current therapies are directed at decreasing vascular resistance through either augmenting vasodilator activity or inhibiting the activity of vasoconstrictor substances. Adiponectin is known to have direct vasodilator activity[47–49] and adiponectin deficiency is associated with the development of systemic hypertension and impaired vasodilation.[50] Adiponectin-deficient mice have also been found to have reduced levels of endothelial cell nitric oxide in the vascular wall and develop an age-dependent increase in pulmonary artery pressure when compared with wild-type mice.[51] These data strongly suggest that adiponectin deficiency may be associated with impaired vasoreactivity; however, there are no studies specifically examining the effects of adiponectin-deficiency on the vasoreactivity of the pulmonary vasculature.
Adiponectin is a suppressor of inflammation
There is a growing appreciation that pulmonary vascular inflammation is an important stimulus for the pathologic changes seen in various types of PAH in both human and animal models.[1,3,46,52] A role for inflammation in the pathogenesis of PAH is suggested by studies demonstrating the presence of increased levels of cytokines in patients with PAH[53,54] and the accumulation of macrophages and T cells in and around the remodeled vasculature of the lung.[55–57] In addition, a recent study has demonstrated a strong correlation between cytokine levels and survival in PAH.[58]
It is believed that chronic low-grade inflammation, as occurs in obesity, contributes to the development of PAH through promoting vascular remodeling. Activation of inflammatory pathways has been shown to stimulate endothelial cell activation, to promote pulmonary artery smooth muscle proliferation and to activate antiapoptotic pathways.[59,60] Indeed, the ability of inflammation to directly induce PAH has been demonstrated in animal models in which chronic inflammation alone was shown to promote pulmonary vascular remodeling and lead to elevated pulmonary artery pressures.[61–65] Overall, these studies support the hypothesis that inflammation is an important component of the pathogenesis of PAH and suggest that processes that suppress inflammation could have a therapeutic role in treating this disease.
One important function of adiponectin is to tonically suppress vascular inflammation. This is exemplified in adiponectin-deficient mice, which develop a spontaneous phenotype characterized by activated lung endothelium, age-dependent increases in perivascular inflammatory cell infiltration and elevated pulmonary artery pressures.[5] In addition, these mice develop an exaggerated eosinophilic vascular response to allergic lung inflammation, which is associated with increased pulmonary artery pressure and muscularization of the pulmonary vasculature.[4] Interestingly, elimination of eosinophils in this model prevents the development of PAH in the adiponectin-deficient mice.[6] Thus, findings in these studies provide further support for the concept that inflammation is an important stimulus for PAH and that adiponectins ability to modulate inflammatory responses may be influential in the development of this condition.
Adiponectin is a suppressor of growth and proliferation
As previously discussed, a characteristic pathological feature of PAH is narrowing and/or obliteration of the vessel lumen due to thickening of the vascular wall. In large muscular arteries, this is usually secondary to medial hypertrophy, while smaller pulmonary arteries and precapillary vessels can be obliterated by plexiform lesions.[46] These changes have been attributed to increased proliferation and migration of mesenchymal cells that stain positive for a-smooth muscle cell actin (α-SMA), indicating that there is dysregulation of local smooth muscle cells (SMCs) or myofibroblast growth.[1] Alternatively, these cells could derive from circulating progenitor cells, but data supporting this hypothesis is less robust.[66,67] Regardless of the cellular origin, these cells are presumably stimulated to divide in response to mitogenic stimuli.
Currently, the particular mitogenic substances that are most important in mediating the progression of PAH have yet to be identified. However, clinical studies have shown that concentrations of multiple growth factors are increased in lung biopsy specimens of patients with PAH.[68–72] For example, transcript and protein levels for platelet-derived growth factor (PDGF), epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF) are increased in distal pulmonary arteries of patients with PAH. Moreover, various experimental studies have shown these factors to be important in promoting SMC proliferation and in augmenting resistance through their effects on cell survival.[68–72] While each of these factors is likely to be important, the search for other mitogenic substances that contribute to the development of PAH is ongoing.
Although adiponectin is not a well-described factor in growth regulation, there is a growing appreciation for its effects on tissue remodeling.[73–75] In vitro, adiponectin suppresses vascular SMC proliferation and migration,[76] and in vivo, adiponectin-deficient mice have increased accumulation of SMCs in vessel walls following vascular injury[77] In addition, adiponectin-deficient mice have increased cardiac remodeling with pressure overload and larger infarct size following cardiac ischemia when compared with wild-type mice.[74,78] As mentioned above, adiponectin-deficient animals develop more prominent pulmonary vascular remodeling in the setting of pulmonary vascular inflammation, and similar findings have also been reported in a model of hypoxia-induced PAH. These findings suggest a general and robust effect of adiponectin on pulmonary artery remodeling[79]
The mechanisms mediating adiponectins inhibitory actions on cell proliferation are poorly understood; however, these actions appear to be independent of its effects on inflammation.[7] In a recent study, it was demonstrated that overexpression of adiponectin reduced pulmonary vascular remodeling in an inflammation-induced model of PAH without reducing vascular inflammation.[7] In addition, adiponectin has been shown to directly affect several signaling pathways in SMCs important for cell proliferation and growth.
The intracellular signaling mechanisms that regulate the SMC phenotype are poorly understood, and likely involve several different signaling cascades.[80] Growth factors such as PDGF, EGF and fibroblast growth factor (FGF) stimulate SMC proliferation in part through phosphorylation of PKB (AKT1) and effects on the mTOR pathway, which stimulates cell growth and proliferation.?[81,82] Adiponectin has been shown to inhibit growth factor-mediated activation of mTOR via AMPK activation.[75,83] In addition, adiponectin has been shown to directly bind to growth factors thus modulating their activity by controlling their bioavailability at a prereceptor level.[76,84] Overall, these data suggest that adiponectin may suppress vascular remodeling via a complex set of mechanisms.
The increased numbers of muscle cells seen in pulmonary hypertension are likely derived from existing SMCs that migrate around the vessel, proliferate and then form new muscle.[85,86] For this to occur, the SMC must first dedifferentiate into a highly proliferative phenotype prior to migration and proliferation, and then later differentiate into a contractile phenotype to form new muscle. Transcription factors such as GATA6, MEF2 and the serum response factor–serum response element (SRF-SRE) pathway regulate the SMC phenotype. SRF is a phylogenetically conserved MADs-box transcription factor that binds a 10-base pair DNA sequence (CC[A/T]6GG or CArG box) and mediates the rapid transcriptional response to growth and differentiation signals. SRF is one of the major regulators of SMC growth, migration, survival and differentiation,[87–90] controlling the expression of more than 200 genes, nearly half of which are involved in cytoskeletal dynamics and cellular contractility.[91] Deletion of SRF in mice is lethal,[92] and SMC-specific deletion leads to impaired vascular SMC differentiation in the embryo.[93] Consistent with this, expression of SMC-specific genes, such as SM-myosin heavy chain, SM α- and γ-actin, SM22α, SM-myosin light chain kinase, calponin, α-actinin and smoothelin-A, are downregulated in the absence of SRF.[94,95] Because the SRF-SRE pathway is the major regulator of SMC growth and differentiation,[87,88] factors that affect it are likely to have a profound effect on the differentiation state of SMCs and may influence vascular remodeling in the lung. Recent data has demonstrated that adiponectin can reduce SRF-SRE activity,[7] and thus could work to limit SMC growth and proliferation by effecting the differentiation of SMCs into a proliferative phenotype. These data suggest that adiponectin may also inhibit pulmonary vascular remodeling by modulating the growth phenotype of SMCs.
Adiponectin is a metabolic hormone
Several findings suggest that metabolism may influence the pathogenesis of pulmonary hypertension. Patients with pulmonary hypertension have reduced expression of PPARγ in the lung, a receptor that regulates adiponectin and insulin resistance.[96] In addition, mice with a targeted deletion of PPARγ in SMCs spontaneously develop pulmonary hypertension with muscularization of distal pulmonary arteries.[97] Furthermore, apoE-deficient mice on a high-fat diet develop PAH. It has also been shown that insulin resistance and dyslipidemia are more common in women with PAH and that insulin resistance was associated with worse outcomes in these patients.[98]
Adiponectin is an important modulator of metabolism leading to improved insulin sensitivity and decreased glucose and free fatty acid levels in the plasma. Specifically, adiponectin stimulates β-oxidation and downregulates the expression of mediators of lipid synthesis.[99] Furthermore, in animal models, ectopic expression of adiponectin promotes metabolic function independent of body mass.[100] At least some of the beneficial effects of the PPARγ agonist class of drugs seems to be related to their ability to increase adiponectin levels.[99] In fact, a recent study has demonstrated that the cardioprotective effects of PPARγ agonists are dependent on their ability to increase adiponectin levels.[101] In accordance with this, male apoE-deficient mice on a high-fat diet do not upregulate adiponectin, but develop insulin resistance and PAH.[96] However, female apoE-deficient mice on a high-fat diet had increased adiponectin levels at baseline and did not develop insulin resistance and had less PAH. Treatment of these mice with the PPARγ agonist rosiglitazone (which increases adiponectin levels) attenuates the PAH,[96] suggesting that the link between metabolism and PAH may relate in part to changes in adiponectin levels.
CONCLUSION AND CLINICAL IMPLICATIONS
There is increasing data suggesting that obesity may be a risk factor for PAH independent of its effects on systemic vascular disease and obesity–hypoventilation syndrome.[102] Although the data does not support a primary role for obesity in causing PAH, the data does suggest that the effects of obesity on metabolism and vascular inflammation could contribute to the development of pulmonary vascular remodeling and PAH. Thus, it is possible that obese patients with PAH from other causes could have more rapid progression and more severe disease than lean patients. Furthermore, as PAH is associated with reduced exercise capacity, many patients may become obese overtime due to an inability to exercise and thus could have accelerated disease due to the effects of obesity on the pulmonary vasculature. Clearly, more research is needed on the exact role obesity may have on the pathogenesis of PAH.
Given the known effects of adiponectin on vascular inflammation and remodeling (Fig. 2), it seems likely that relative deficiency of adiponectin as seen in obesity could be an important mechanistic link between obesity and PAH. Animal models demonstrate that adiponectin can modulate pulmonary vascular inflammation and remodeling, which then directly influences the development of PAH. Whether there is a similar effect in humans with PAH is unknown at this time, but the data does suggest that measures to augment adiponectin levels could have therapeutic value in patients with PAH, especially those with obesity and insulin resistance. It should be noted that such therapy is already available with the thiazolidinedione class of antidiabetic drugs (such as pioglitazone), which increase adiponectin secretion by stimulating PPARγ. Based on the available data, a trial of these agents in PAH patients with obesity may be warranted.
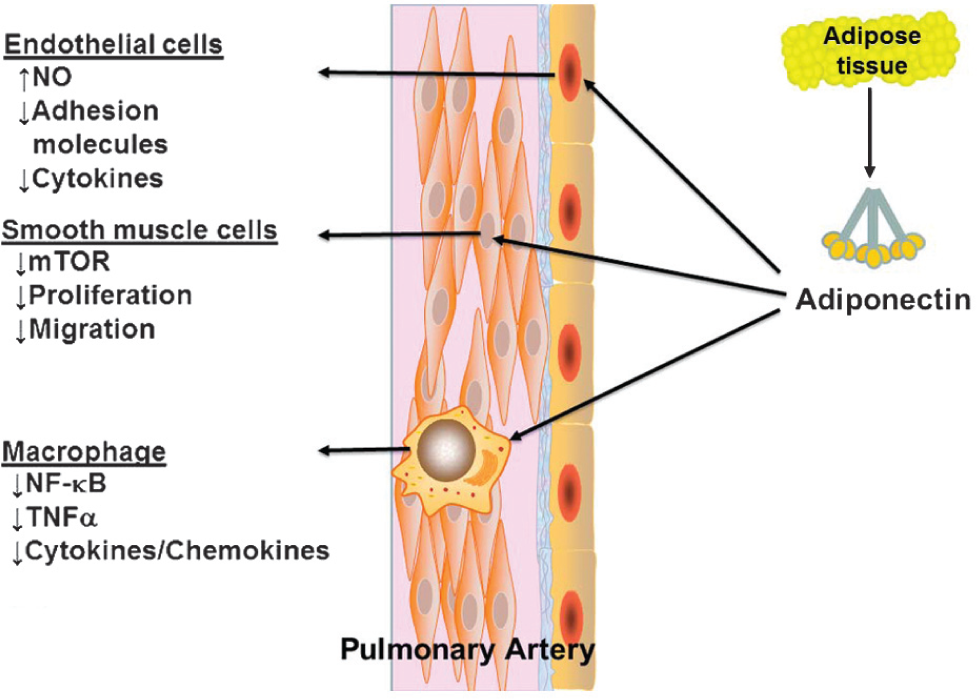
Effects of adiponectin in the pulmonary vasculature.