
Abstract
Pharmacogenomics is the study of how genetic variations influence the response to drugs, by correlating gene expression with the drug's efficacy and toxicity. This concept has recently been successfully applied in oncology. To test its applicability to PAH, we examined two experimental models of the disease: mice with deletion of the Vasoactive Intestinal Peptide gene (VIP-/-); and rats injected with monocrotaline (MCT). Since the two models express comparable phenotypic features, we analyzed their particular gene alterations, with special reference to genes related to pulmonary vasoconstriction, vascular remodeling, and inflammation. We then compared the phenotypic and genotypic responses in each model to treatment with the same drug, VIP. In untreated VIP-/- mice there was over-expression of almost all genes promoting vasoconstriction/proliferation, as well as inflammation, and under-expression of all vasodilator/anti-proliferative genes. As expected, treatment with VIP fully corrected both the key PAH features, and all gene expression alterations. MCT-treated rats showed two distinct sets of alterations. One, similar to that in VIP-/- mice, i.e., tended to promote vascular remodeling and inflammation, e.g., up-regulation of myosin polypeptides, procollagen, and some inflammatory genes. The other was a set of opposite alterations that suggested an effort to modulate the PAH, e.g., up-regulation of the VIP and NOS3 genes. In this model, VIP treatment failed to correct many of the genotypic abnormalities, and, in parallel, incompletely corrected the phenotypic changes as well. This preliminary proof-of-concept study demonstrates the importance of genomic information in determining therapeutic outcome, and thus in selecting personalized therapy. Full validation of the merits of pharmacogenomics must await studies of lungs from patients with different forms of PAH.
Keywords
INTRODUCTION
What is pharmacogenomics?
Pharmacogenomics is the study of how genetic variations influence the response to drugs, through correlation of gene expression or single nucleotide polymorphisms (SNPs), with the drug's efficacy and toxicity.[1,2] By probing the gene expression profile of individual disorders within a broad group of disorders, specific therapy can be targeted to that disorder, leading to greater efficacy and reduced toxicity. This approach has been described as leading to “the path to safer and more effective drugs”,[3] and “the path to personalized medicine”.[4]
The promise of pharmacogenomics is fulfilled in oncology
Over the past decade, the concept of pharmacogenomics has been applied in the field of oncology, often with dramatic success. Notable results have been reported in the treatment of: breast cancer, in relation to ErbB2/HER2 amplification;[2,5] lung cancer, in relation to EGFR mutations;[5,6] and colorectal cancer in relation to EGRF-targeted therapy.[5,7]
Like cancer, PAH is a heterogeneous disorder—should it benefit from application of pharmacogenomics?
As with most solid tumors, which may appear morphologically similar but have different and distinct genomic variations, PAH is a heterogeneous disorder, possibly with multiple different genotypes. It is already widely recognized that different clinical groups of PAH exhibit different degrees of responsiveness to the same therapeutic agents, with PAH associated with scleroderma and other inflammatory disorders having a generally poorer response than idiopathic PAH.[8] Such differences in therapeutic response probably reflect differences in underlying gene expression patterns, which, therefore, may benefit from targeted therapies based on unique genomic features.
Hypothesis
This preliminary study is designed as a first step in validating the hypothesis that PAH, much like cancer, is sufficiently diverse as to merit the application of pharmacogenomics, i.e., gene-targeted therapy based on the specific genetic background of each individual case. Such personalized therapy should lead to enhanced survival and minimized side effects.
MATERIALS AND METHODS
Animals
Vasoactive Intestinal Peptide (VIP) knockout mice (VIP−/“), backcrossed to C57BL/6 mice,[9] were bred locally as described (10), and genotyped to confirm the absence of the VIP gene.[10] Sprague Dawley (SD) rats, 200-230 g, were from Taconic Labs (Germantown, NY). All experiments and animal care procedures were approved by the Institutional Animal Care and Use Committee and were conducted according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Chemicals and reagents
Monocrotaline (MCT) was from Sigma-Aldrich (St. Louis, Mo.), and VIP was from Bachem Americas Inc. (Torrance, Calif.).
Study design: Validating pharmacogenomics in experimental models of PAH
As an initial effort to test the applicability of pharmacogenomics to PAH, we examined two experimental models of PAH: male VIP7’ mice, 10-12 weeks old; and male 6-8-week-old rats injected with MCT (60 mg/kg) 3 weeks earlier. These two models express comparable phenotypic features, with the MCT model showing generally more severe PAH.[10,11]
We analyzed and compared the genetic alterations in the lungs of animals from both models, with special reference to genes directly related to promoting or modulating of pulmonary vascular remodeling or inflammation, and to the extent to which these abnormalities were corrected in response to treatment with the same drug, VIP.
Experimental groups
The mouse groups were: 5 untreated VIP−/− mice; 5 VIP−/− mice, treated with VIP (500 ng/kg, i.p., every other day for 3 weeks, for a total of 10 injections); and 5 normal wild-type mice. The rat groups were: 5 SD rats injected with MCT 3 weeks earlier, not otherwise treated; 5 SD rats injected with MCT, then treated with VIP, (500 μg/kg, i.p., every other day, for 3 weeks), beginning 3 weeks after MCT; and 5 normal SD rats, not injected with MCT or VIP.
Statistical analysis
Differences in quantitative data among the experimental groups were analyzed by ANOVA and Tukey post-hoc tests for multiple comparisons. Unpaired students t-test were used to analyze differences between two animal models. A P value of > 0.05 was considered significant.
Measurements
Both the mouse groups and the rat groups were compared with regard to: phenotypic features of PAH (RV systolic pressure, RV hypertrophy, pulmonary vascular remodeling, lung inflammation); gene expression alterations related to PAH; and the degree to which treatment with VIP corrected phenotypic and genotypic alterations.
Hemodynamic measurements
Animals were anesthetized with ketamine (100 mg/kg) and fentanyl (0.05 mg/kg IP). A 1.4F 3-cm Mikro-Tip catheter (Millar Instruments Inc, Houston, Tex.) was inserted through the right jugular vein and advanced to the right ventricle for digital recording of RV pressure.
Histological examination and morphometric analysis
For all histological procedures, the lungs were inflated to full capacity and fixed by intratracheal instillation of 10% neutral buffered formalin, immersed in formalin overnight, and then embedded in paraffin. Sections (4 μm thick) were stained with hematoxylin and eosin or Masson's trichrome stain for general morphology and morphometric analysis. Pulmonary arteries were analyzed; measurements were taken of 4 separate vessels from each slide and averaged to 1 set of values. Only arteries near smaller bronchi or terminal bronchioles, 50 μm in diameter, were selected for analysis. The Image J program, version 1.34r (http://rsb.info.nih.gov/ij/), was used for measurement of total vessel area (mm2), luminal area (mm2), and inner circumference (mm). Medial area (mm2) was calculated as the difference between total and luminal areas. Standard medial thickness (mm) was calculated as the ratio of medial area to inner circumference, as described.[12]
Anatomic assessment of RV hypertrophy
The heart was isolated and placed under a dissecting microscope. Attached vessels and both atria were dissected and removed. The RV wall was cut out, blotted, and weighed; then the left ventricular wall and septum (LV+septum) were treated the same way and weighed. The RV/(LV/septum) ratio was calculated as an index of RV hypertrophy.
Assessment of lung inflammation
Lung sections were examined by a pathologist who was blinded to the identities of the samples. Inflammation was graded 0, 1, 2, 3, or 4, based on the intensity and extent of perivascular and peribronchiolar inflammatory cell infiltrates.
Microarray and RT PCR analysis
Lungs from mice and rats were removed from freshly euthanized animals, immersed in RNA later TM (10 ml per mg of tissue; Ambion, Austin, Tex., USA), fresh-frozen in liquid nitrogen and shipped overnight on dry ice to SA Biosciences (Qiagen, Fredrick, Md., USA). Microarray data were collected using the Whole Mouse Genome Oligo Microarray Kit with SurePrint technology (4644K slide format; Agilent Technologies, Palo Alto, Calif., USA). The microarray results were validated using quantitative real time PCR. RNA was examined from the same lungs subjected to microarray analysis. The procedures were carried out and the results were analyzed at Superarray Biosciences. Gene expression data of lungs from the mouse and rat groups were separately analyzed relative to un-treated animals, according to the 2 −AAC T method.[13]
RESULTS
Both models expressed the key phenotypic features of PAH, including RV systolic pressure, pulmonary vascular smooth muscle, right ventricular (RV) hypertrophy, and lung inflammation, with the MCT syndrome being generally more severe (Table 1), and uniformly fatal within 3-5 weeks.
Phenotypic features in MCT rats and VIP−/− mice models of PAH

Measured as medial area/total area in pulmonary arterioles 50-75 μM diameter.
Measured as RV/(LV+Septum) weight ratio
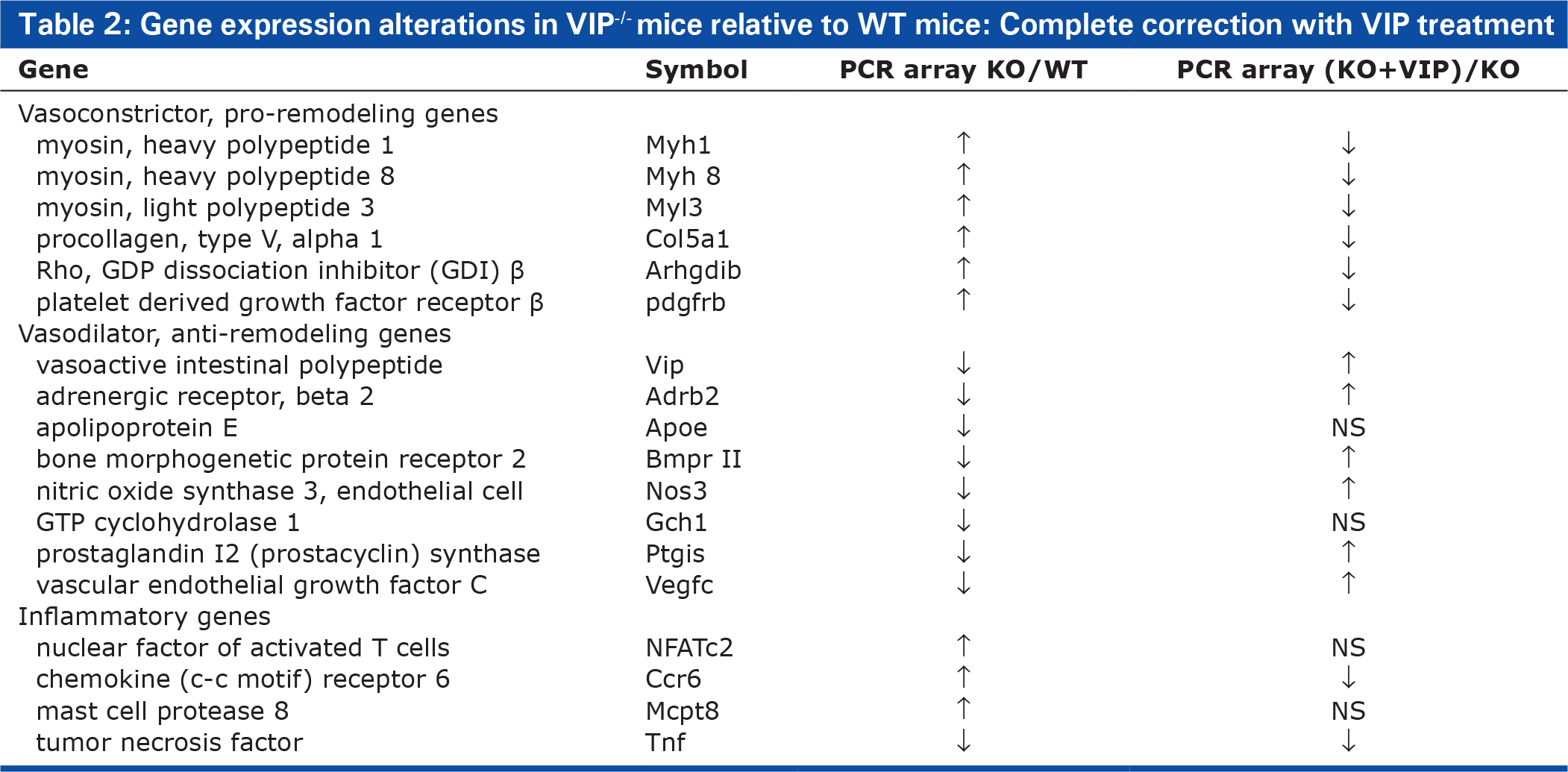
Lungs from VIP KO mice uniformly expressed a single set of gene alterations, comprising: up-regulation of vasoconstrictor/proliferative genes, including myosin heavy and light polypeptides, procollagen peptides, Rho-related genes, angiopoetin, and platelet-derived growth factor receptor; down-regulation of vasodilator/anti-proliferative genes, including adrenergic receptor β, GTP cyclohydrolase 1, the rate-limiting enzyme in the biosynthesis of tetrahydrobiopterin (BH4), an essential co-factor for the activity of endothelial nitric oxide (NO) synthase (eNOS or NOS3), prostacyclin synthase, apoplipoprotein E, and adrenomedullin receptor; and up-regulation of inflammatory genes, including tumor necrosis factor, mast cell protease 8, chemokine (C-C motif) receptor 6, and NFATC2 (Table 2). Treatment of these mice with VIP corrected the key features of PAH, including the elevated pulmonary artery pressure, vascular remodeling, right ventricular hypertrophy, and lung inflammation. As well, the treatment reversed the genetic alterations toward normal values (Table 2).
Gene expression alterations in VIP−/− mice relative to WT mice: Complete correction with VIP treatment
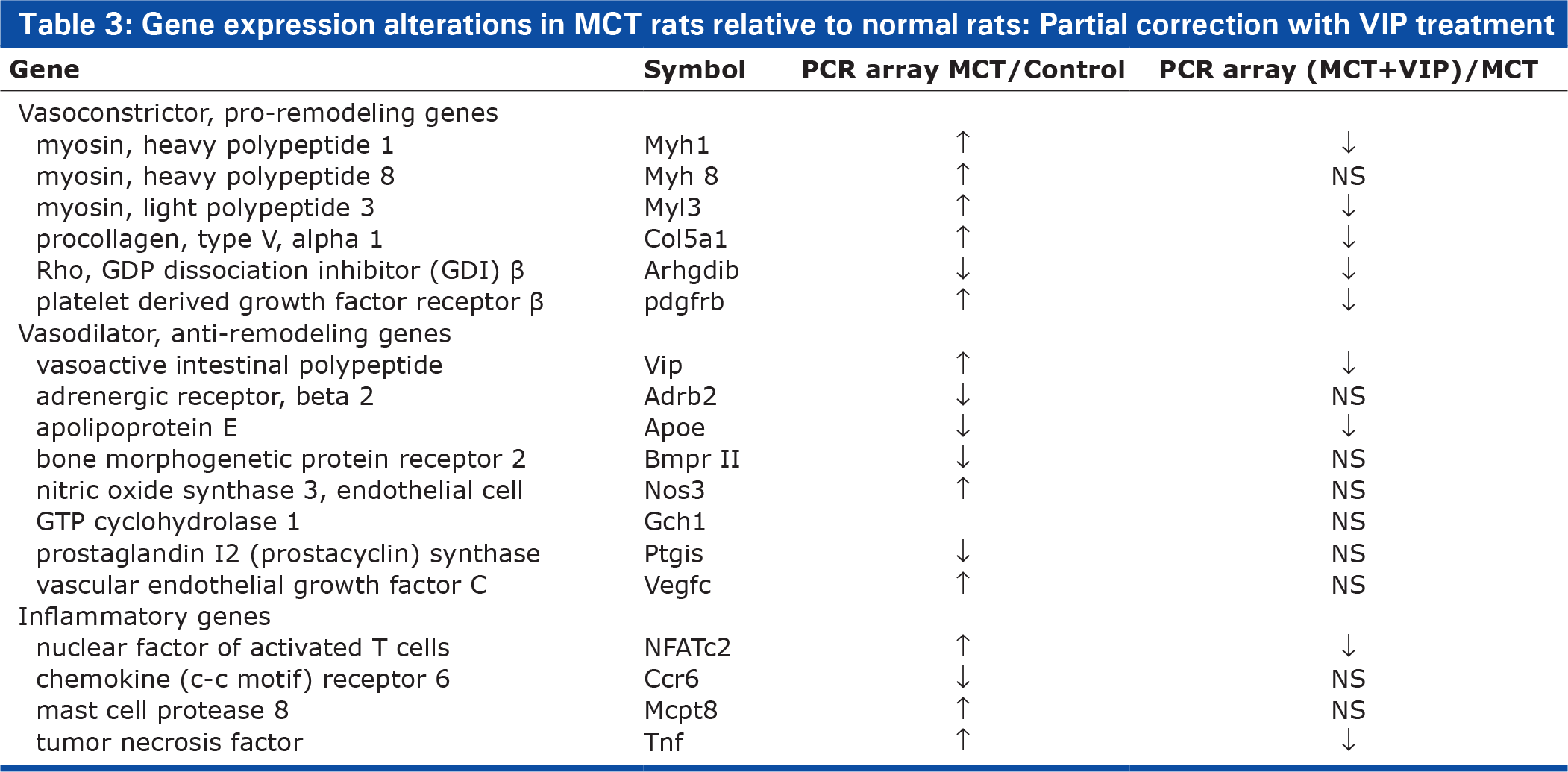
With respect to the MCT model, however, VIP treatment incompletely corrected the phenotypic changes: it significantly but only partially attenuated the pulmonary hypertension, vascular remodeling, and lung inflammation, but only slightly and insignificantly reduced RV hyperterophy. Following VIP treatment, genotypic analysis showed two distinct sets of expression alterations: one, similar to that in VIP'−/− mice, i.e., alterations that promote vascular remodeling and inflammation (e.g., up-regulation of myosin polypeptides, procollagen, and of some inflammatory genes); and another set of alterations that suggested an effort to modulate the PAH. (e.g., up-regulation of VIP and NOS3). As with the phenotypic abnormalities, VIP treatment was only partially successful in reversing the genotypic abnormalities (Table 3).
Gene expression alterations in MCT rats relative to normal rats: Partial correction with VIP treatment
DISCUSSION
As noted above, the objective of this study was to provide preliminary evidence in support of applying the concept of pharmacogenomics in the treatment of PAH. We chose two experimental models of the disease that share major pathophysiologic and pathologic features, though to different degrees of severity, thus mimicking the groups of PAH patients with the same overall clinical diagnosis. The two models have sharply different survival rates, with VIP−/− mice surviving approximately 15 months[10] and MCT rats only 4-5 weeks, probably because: MCT is poisonous to several vital organs, including the lungs, liver, and kidneys; absence of the VIP gene results in two lung disorders (PAH, an asthma-like condition, possibly with altered function of other organ systems, but none rapidly fatal); and lack of the VIP gene is at least partially compensated for by the presence of two closely related VIP-like peptides (the Pituitary Adenylate Cyclase Activating Peptides, PACAP-27 and PACAP-38).
Our plan was: to analyze gene expression alterations in the two models, with special reference to genes related to pulmonary vasoconstriction, vascular remodeling, and inflammation; to compare the phenotypic responses of the two models to treatment with one and the same drug, VIP, which seems particularly suited to the genetic background of one model (VIP−/− mice), but not to the other (MCT rats); and to correlate the success of therapy in reversing phenotypic abnormalities with the success in correcting the corresponding genotypic alterations.
VIP therapy was more successful in one model than the other
Both animal models examined were treated with the same dose of VIP per weight, injected i.p. We did not measure serum level of VIP following VIP treatment. This decision was based on earlier experience with successful treatment of VIP−/− mice with VIP,[10] and the facts that VIP has a very short half life in serum or plasma, and its actions are mediated chiefly by binding to receptors in key cells and tissues, which would not be reflected in serum levels.
The fact that deletion of the VIP gene alone resulted in expression of the PAH phenotype, together with marked alterations in the expression of numerous genes that promote the development and aggravation of PAH, clearly identifies the underlying mechanism of pathogenesis of PAH in that model. The additional fact that the administration of VIP fully corrected both phenotypic and genotypic abnormalities is further proof of this conclusion.
In this case, therefore, with the knowledge that PAH was strictly attributable to absence of the VIP gene, and the consequent gene expression alterations, it was predictable that VIP treatment would fully correct both phenotypic and genotypic abnormalities.
In the MCT rats, however, since the VIP gene was not under-expressed, it was evident that VIP deficiency was not the primary defect in the MCT model, and thus VIP treatment alone, unlike in the VIP−/− mice, could not be expected to correct either the genotypic or phenotypic abnormalities. The complex and mixed nature of the genotypic picture strongly indicates that effective therapy required multiple therapeutic approaches.
Relationship to gene expression alterations in human PAH
A number of the gene expression abnormalities noted in the VIP−/− mice or the MCT rats resemble those reported in human PAH. These include the following.
Reduced BMPR-II expression is a feature in common between both models and of the non-Familial forms of PAH.[14]
One consequence of reduced BMPR-II function is the increased expression of pro-inflammatory cytokines and inflammation[14,15] An inflammatory response is a key feature of human PAH, as well as of the 2 models we examined[16–18]
Numerous other examples exist of shared gene expression abnormalities between the human disease and either or both of the animal models. These include increased expression of vasoconstrictor, pro-remodeling genes, e.g., angiopoetin and decreased expression of apolipoprotein E, and of voltage-gated potassium channels.[17–22]
The potential importance of VIP gene mutation in human PAH has not been adequately examined. Two clinical trials have been conducted with VIP in human subjects with PAH. In one trial, the peptide, given to 8 patients with IPAH by inhalation, for up to 24 weeks, resulted in significant improvement in hemodynamics and exercise tolerance.[23] In a more recent study, reported only in abstract form,[24] the results were described as negative. In neither of these trials was any information available on genomic data.
CONCLUSIONS
Despite its limited scope, this proof-of-concept study provides evidence supporting the applicability of the concept of pharmacogenomics in PAH. The two examples of correlating genomic analysis with drug response demonstrate the usefulness of genomic information in selecting appropriate, targeted therapy in PAH, and predicting therapeutic outcome. Complete validation of the importance of pharmacogenomics in human PAH must await similar studies in lung tissues from patients with different forms of PAH. Such tissues, likely from patients undergoing lung transplantation, are available from the Pulmonary Hypertension Breakthrough Initiative (PHBI), at the University of Michigan, Ann Arbor, Michigan, USA. A clinical trial to confirm the importance of pharmacogenomics in human PAH is already underway.[25]