
Abstract
Pharmacotherapeutic options for pulmonary arterial hypertension (PAH) have increased dramatically in the last two decades and along with this have been substantial improvements in survival. Despite these advances, however, PAH remains a progressive and ultimately fatal disease for most patients and only epoprostenol has been shown to improve survival in a randomized control trial. Clinical observations of the heterogeneity of treatment response to different classes of medications across the phenotypically diverse PAH population has led to the identification of patients who derive significantly more benefit from certain medications than the population mean, the so-called “super responders.” This was first recognized among PAH patients with acute vasodilator response during invasive hemodynamic testing, a subset of whom have dramatically improved survival when treated with calcium channel blocker (CCB) therapy. Retrospective studies have now suggested a sex discrepancy in response to endothelin receptor antagonists (ERA) and phosphodiesterase inhibitors, and more recently a few studies have found genomic associations with response to CCBs and ERAs. With increasing availability of “omics” technologies, recognition of these “super responders,” combined with careful clinical and molecular phenotyping, will lead to advances in pharmacogenomics, precision medicine, and continued improvements in survival among PAH patients.
Pulmonary arterial hypertension (PAH) is a disease characterized by progressive pulmonary vascular obliteration and remodeling that leads to increased pulmonary vascular resistance (PVR) and ultimately right heart failure and death.1–4 PAH is seen in idiopathic (IPAH) and heritable (HPAH) forms, with an increasing number of genes now known to contribute to predisposition to the disease.5,6 PAH can also be see in association with drugs or toxins, or other diseases such as connective tissue disease (CTD), Human Immunodeficiency Virus (HIV) infection, portal hypertension, congenital heart disease (CHD), and schistosomiasis.2,7
Drugs for the treatment of PAH and predictors of response to therapy.

Not FDA-approved for use in PAH.
Two factors leading to these less encouraging results may be heterogeneity of treatment response and patient selection for clinical trials. In order for a drug to obtain FDA approval, it must demonstrate safety and effectiveness on average in the studied population versus placebo or usual care. When examining the effectiveness of a particular drug, all patients enrolled in the trial are taken into consideration, and the mean change in desired outcome, be it survival, time to clinical worsening, or six-minute walk distance (6WMD), is generally interpreted as the most significant factor in determining whether that therapy should be implemented into practice. Regardless of the mean outcome, however, there may be patients within the population that respond remarkably well to therapy, so-called “super responders,” while others have no response, or even adverse response to the same treatment.
On one hand, it is valuable to have broad entry criteria to facilitate more rapid and wide patient enrollment into trials. However, including patients that may be less likely to respond to a drug, such as CTD patients, who are less likely to have improvements in clinical parameters such as 6MWD and FC due to musculoskeletal limitations, also substantially increases the heterogeneity of response, and the beneficial effects seen in a subset of the population may be lost in the final analysis.21,22
We are entering an era of precision medicine, one in which algorithm-based treatment approaches will be modified in ways that take individual variability into account. 23 We have already seen great strides using this approach in oncology, such as the use of CTLA-4 blockade in melanoma and ALK inhibitors in lung cancer.24,25 The pulmonary field has also seen early advances in precision medicine, specifically in cystic fibrosis (CF) and the approval of ivacaftor for patients with the G551D mutation. 26 This provides an excellent example of a therapy that, if applied across the whole of the CF population, would be unlikely to have a net positive clinical effect and may not have garnered FDA approval, but targeted specifically to the 4–5% of CF patients that harbor the G551D mutation, resulted in substantial improvements in CF exacerbation rates, symptoms, weight, and lung function. 26
The genetics of PAH, like CF, have been studied for decades, and we now know of many genes that predispose some amount of risk for the development of PAH since the original discovery of BMPR2 in HPAH.5,6,27 Despite this increasing knowledge of genetic predisposition, however, there are presently no PAH therapies targeting these genes or their products, and patients’ individual molecular etiology plays no role in the selection of PAH directed therapy, which remains algorithm-based and driven by severity of disease. 28 With the availability and increasing affordability of high-throughput “omics” technologies, and careful phenotyping of patient cohorts, soon it may be possible to use these techniques to identify patients more likely to respond to current PAH therapies, as well as develop novel targets for future therapies.29,30
The need for targeted PAH therapy, and appropriate selection of initial pharmacotherapy, can be seen in examples from multiple clinical trials of PAH medications, where it appears that even short-term treatment with placebo appears to portend long-term negative consequences.31,32 These patients, who received placebo for as short as 12 weeks, often do not reap the same benefits as those in the active treatment arm, even after long-term follow-up in the open-label extensions of the trials. 32 In addition to posing important questions about trial design for future PAH therapies, this emphasizes the importance of appropriate, and ideally patient-specific, drug selection upfront for PAH patients, so that clinical deterioration does not occur in the setting of ineffective therapy, knowing that the ground lost quite possibly will not be recovered later, when therapy is changed.
The current treatment paradigm for PAH, after establishing a definitive diagnosis via right heart catheterization (RHC) with acute vasodilator response (VR) testing, and ruling out alternative causes for pulmonary hypertension, hinges on establishing a patient’s risk for immediate adverse outcomes to guide pharmacotherapy. 2 Signs of right heart failure, tempo of disease, syncope, New York Heart Association (NYHA) or World Health Organization (WHO) functional class, and further testing such as 6MWD, cardiopulmonary exercise testing (CPET), brain natriuretic peptide (BNP) or N-terminal proBNP (NT-proBNP) levels, echocardiographic assessment of right atrial (RA) size, presence of pericardial effusion, and invasive hemodynamic assessment of right atrial pressure (RAP), cardiac index (CI), and central venous oxygen saturation (SvO2) are all used to recommend treatments. 2 Following this determination of functional class and risk, the most recent guidelines list the indicated drugs for each functional class and the strength of recommendation. While these guidelines serve as an invaluable resource and compilation of the available evidence for treatment of PAH, clinicians are still left with the difficult task of selecting what they believe will be the best treatment option for the patient in front of them, selecting from the ten drugs or drug combinations with level 1 evidence for WHO FC II and III. 2
The evidence to guide clinicians in this vital decision is limited at present, with retrospective data suggesting that there may be disparities in response to certain drugs or drug classes by PAH subtype, sex, or race, and two studies that have begun to look at molecular predictors of response to PAH specific therapy, which have not yet been applied at the bedside.33–37 Despite the paucity of clinical trial data to guide these decisions, clinicians have recognized the phenomenon of “super responders” to PAH medications; patients who have sustained clinical improvement to a certain pharmacotherapy, in contrast to patients who progress to two or three drug combinations with continued decline. This is a phenomenon also seen in oncology, wherein a small number of treated patients may have complete remission and/or prolonged survival despite advanced disease. 38 Further investigation into characterizing these “super responders,” and development of predictive tools prior to selection of therapy, could have a major impact on patient care in many diseases, and will be increasingly feasible with the emerging clinical availability of molecular (e.g. genomic, proteomic, and metabolomic) technologies.
This review will focus on what is presently known about predictors of response to PAH pharmacotherapy, the identification of “super responders” to these therapies, and future directions for tailoring the available PAH drugs to the patients most likely to benefit from them. There is no consensus definition of “super responders” in PAH at this time, a challenge unto itself, so we will focus primarily on the identification of patients who, when treated with a certain medication, have prolonged survival compared to otherwise matched patients, and those who have significant and sustained improvements in clinical parameters such as 6MWD, time to clinical worsening, or functional class when compared to similarly treated patients.
Calcium channel blockers
Years before epoprostenol was synthesized, trialed, and approved for the treatment of PAH in the 1990s, there was recognition that a subset of PAH patients had substantial clinical improvement with vasodilator therapy, and specifically CCBs.9,39,40 After initial early attempts at treating PAH with various vasodilators, with mixed results, it was the close observation of relatively few patients with careful clinical phenotyping that allowed Rich et al. to define the subset of PAH patients that responded well to high-dose CCBs.40–42 Initially the criteria to identify these patients were imprecise and the methods of performing vasodilator response testing varied across institutions, resulting in discrepancies with reported rates of acute vasodilator responsiveness and long-term CCB responders. Over many years of experimentation and observation, however, acute VR testing was honed, and by carefully examining the hemodynamic changes during VR testing in hundreds of patients with long-term follow-up data, the hemodynamic criteria for identifying candidates for CCB treatment became much more precise. 10 Presently, acute VR testing is a class 1 recommendation in all newly diagnosed patients with IPAH, HPAH, or drug-/toxin-related PAH, and the criteria for a positive response is reduction in pulmonary artery pressure (PAP) ≥ 10 mmHg to reach an absolute value of mean PAP ≤ 40 mmHg with an increased or unchanged cardiac output. 2 These criteria, as of now, provide the best opportunity to identify the 5–10% of PAH patients who benefit from long-term CCB therapy. The reason it remains so important to identify this subset of patients, even in the modern treatment era with ten new available drugs, is that they represent the original “super responders” to PAH treatment. In the paper that most clearly defined the criteria for long-term CCB response, Sitbon et al. reported that with mean follow-up of seven years, all but one (98%) of the long-term CCB responders were alive and FC I or II with sustained hemodynamic improvement, whereas among non-responders, the five-year survival rate was 48%. 10 Unfortunately, this seems to apply to only IPAH, HPAH, and drug-/toxin-related PAH patients, with no clear benefit to long-term CCB therapy seen in associated forms of PAH, and uncertain benefit of partial or “non-classic” vasodilator response.43–45
With such a profound clinical phenotype, a molecular basis for this difference between long-term CCB responders and non-vasodilator responsive PAH patients seemed likely. This has been the subject of recent work by our group, first by examining RNA expression patterns of cultured lymphocytes and peripheral blood in CCB responders compared with non-responders. 36 We found 13 genes that were significantly different between the two groups in quantitative polymerase chain reaction (qPCR) analysis of peripheral blood, including cytoskeletal/rho-GTPase genes, cell–cell adhesion genes, developmental genes, and transcription factors, three of which were known to be calcium dependent or activated by adenosine. 36 Furthermore, we were able to design decision trees based on these differentially expressed genes that reliably detected CCB-responsive PAH patients from non-responders in two patient cohorts. 36 In another study, we performed whole exome sequencing comparing CCB responders and non-responders, identifying 1369 genes with 1580 variants unique to IPAH, which identified differences in biologic pathways such as cytoskeletal function, ion binding, and Wnt signaling, and showed enrichment in genes related to vascular smooth muscle contraction. 46 The significance of this work is not only in discovering potentially different molecular causes of the two PAH phenotypes, but also providing a less invasive and reliable means of identifying CCB responders, opening the doors to precision medicine and pharmacogenomics in PAH.
Endothelin receptor antagonists
Increased endothelin (ET-1) activation has been demonstrated in plasma and lung tissues in all forms of PAH, and although it is unclear if this association is a cause or a consequence of the disease, the role of the ET-1 system in PAH pathogenesis, via vasoconstriction and mitogenicity, has been well established.47,48
Endothelin receptor antagonists (ERAs) have been a commonly used first-line agent in the treatment of PAH since the FDA approval of the dual endothelin receptor (ETA and ETB) antagonist bosentan in 2001. This was a major breakthrough in the treatment of PAH, as the first oral therapy proven to be effective in a large-scale RCT. 49 Since then, two additional ERAs have been approved: the selective ETA antagonist ambrisentan and the dual ERA macitentan.19,50
Clinically, marked variation in response to ERAs is seen in PAH patients, with some having significant and sustained improvement in hemodynamic and clinical parameters, while others seemingly have little or no response, or adverse events requiring cessation of the drug and seeking alternate therapy. Some evidence of this heterogeneity of treatment response can be seen in the original phase II trial of bosentan, where despite its proven efficacy across the treatment population as a whole (n = 32), fewer than half of the treated patients had improvement in functional class at 12 weeks. 51 The one-year follow-up data from this trial also support this heterogeneity of treatment response, with some patients displaying dramatic decreases in PVR, while others had minimal change and one patient had an increase. 52 Similarly, three patients had significant increases in cardiac index, perhaps representing “super responders” to this therapy, while others had a more modest increase, or even drop at one year. 52
Unfortunately, little is known about how to predict which patients will respond to ERAs favorably. There is known variation in circulating ET-1 plasma levels according to sex and race from the systemic hypertension literature, leading researchers to wonder whether these factors could play a role in response to ERAs in PAH patients.53,54 Gabler and colleagues retrospectively analyzed the data from six RCTs of ERAs in PAH and compared the outcomes of patients by sex and race; they found that the mean placebo-adjusted treatment response in women was improvement in 6WMD by 44.1 m, compared to 16.7 m among men, a statistically significant difference. 33 They also found that the mean placebo-adjusted treatment response among whites was increase in 6WMD by 41.5 m, whereas in blacks 6WMD decreased by 3.5 m, though this finding did not quite reach statistical significance, owing to the small number of blacks enrolled in the trials. 33 This is an important finding that could impact the treatment of PAH patients and selection of initial therapy, and further suggests the possibility of genetic or hormonal differences playing a role in response to these drugs.
Benza and colleagues took the examination of heterogeneity in response to ERAs one step further, investigating specific ET-1 pathway polymorphisms and how they affected response to ERAs and subsequent clinical outcomes. 35 They selected candidate ET-1 pathway genes from a previously performed genome-wide association study (GWAS) on 715 patients of European descent in the STRIDE trial and found a variant in a G-protein component (GNG2) that significantly increased the rates of developing improvement in 6MWD. 35 The minor allele of this gene variant has a frequency of approximately 12.67%, thus making it a relatively common polymorphism, that correlates with improved response to ERA therapy. While this finding will need replication and prospective validation before it can be incorporated into routine clinical use, it provides an exciting step towards identifying “super responders” to conventional PAH therapy and incorporating pharmacogenomics into the treatment of these patients. 55
Phosphodiesterase type 5 inhibitors
Phosphodiesterase type 5 (PDE5) inhibitors were the second class of orally available agents proven to be efficacious in PAH and have been commonly used as first-line therapy since the FDA approval of sildenafil for PAH in 2005. PDE5 inhibitors slow degradation of cyclic guanosine monophosphate (cGMP), resulting in vasodilatation through the nitric oxide (NO)/cGMP pathway, and also have anti-proliferative effects. 56 Sildenafil, and later tadalafil, have both been proven to increase exercise capacity, and tadalafil has also been shown to increase time to clinical worsening.57,58 Despite widespread clinical use in both PAH and erectile dysfunction, however, little research has been done into predictors of clinical response to PDE5 inhibitor therapy. Mathai et al. retrospectively analyzed data from the PHIRST trial, a RCT of tadalafil in PAH, to identify baseline characteristics that were predictive of achieving clinical improvement on therapy.34,58 Using multivariable logistic regression analyses, they found that men had significantly greater odds of achieving the minimal important difference (MID) in 6MWD. 34 They also found that younger age, lower baseline 6WMD, and IPAH or HPAH (as opposed to CTD-PAH) were associated with greater odds of achieving the MID in 6WMD. 34 This is in an interesting juxtaposition to the findings by Gabler et al., who found that women had greater clinical response to ERAs than men, and if these findings were replicated, they may offer some initial guidance for preferential first-line treatment for FC II-III PAH patients based on sex. 33 A similar retrospective analysis of the PHAROS registry by Lammi et al. also suggested that among patients with systemic sclerosis-related PAH, initial PDE5 or PDE5/ERA combination therapy resulted in improvement in time to clinical worsening compared with initial ERA monotherapy. 37 Unfortunately for now there are no known molecular predictors of response to PDE5 inhibitors and little is known about predicting which patients will have supra-normal response to these drugs, but this would be an interesting area of further research.
Guanylate cyclase stimulators
Like PDE5 inhibitors, soluble guanylate cyclase (sGC) stimulators act to enhance the NO-cGMP pathway, a critical pathway in the pathogenesis of PAH, resulting in vasodilatation and anti-proliferative effects.59,60 Riociguat, the only commercially available sGC stimulator, was FDA approved for the treatment of PAH and chronic thromboembolic PAH following landmark RCTs in 2013, where it was found to significantly increase the 6MWD in both groups.29,60 Like ERAs and PDE5 inhibitors, riociguat has a class I recommendation for the treatment of FC II–III treatment-naïve PAH patients, and perhaps even more so than ERAs and PDE5 inhibitors, little is known about specific predictors of response to therapy. What is known comes from analysis of the PATENT-2 long-term extension trial and, not unsurprisingly, 6WMD, WHO functional class, and NT-proBNP levels at baseline and after 12 weeks of therapy were significantly associated with time to clinical worsening and long-term survival. 61 Similar findings have been seen in large patient registries, regardless of pharmacotherapy.13,62 While helpful in establishing prognosis in PAH patients as a population, this offers little insight to clinicians charged with the task of choosing a first-line agent for PAH patients, and presently we have no way of predicting which individual patients may be “super responders” to riociguat and which should be treated with alternative drugs or initial combination therapy.
Prostacyclin analogues
Prostacyclin is an endogenous compound, produced primarily by endothelial cells, that induces potent vasodilatation in all vascular beds and has cytoprotective, anti-platelet, and anti-proliferative properties. 63 It is a lipid-derived molecule, generated by the cleavage of arachidonic acid by cyclooxygenase and prostacyclin synthase, and signals primarily through the G-Protein coupled IP receptor, resulting in increased cAMP and vasodilatation, but also has important downstream effects on peroxisome proliferator-activated receptor-γ (PPARγ) that may play a role in its anti-proliferative properties. 64 Dysregulation in prostacyclin metabolic pathways has been long established in PAH, with perturbations in prostacyclin synthase, IP receptor expression, and decreased excretion of prostacyclin metabolites in PAH patients.65–67 Because of this, the prostacyclin pathway was one of the first directly targeted therapies for PAH, and synthetic prostacyclin, in the form of intravenous (i.v.) epoprostenol, was the first FDA-approved treatment for PAH.20,68 In addition to being the first FDA-approved therapy, i.v. epoprostenol remains the most efficacious, with years of data from trials and registries showing improved survival in IPAH, CHD-PAH, and CTD-PAH patients.17,69,70 It is the only medication with a class I recommendation for WHO IV patients, is indicated for any PAH patient with evidence of right heart failure or progression of disease despite oral therapy, and has been repeatedly shown to improve right ventricular function.2,71 Despite being the most efficacious treatment option for PAH, however, there remains heterogeneity of response to i.v. epoprostenol, with some patients having long-term survival after being rescued from WHO IV functional class, while others fail to improve and progress to right heart failure, death, or transplant despite therapy. 72 It has even been reported that some patients may have near normalization of hemodynamics, and can later be weaned off i.v. epoprostenol to oral therapies. 73 Failure to respond to i.v. epoprostenol, however, is an independent risk factor for mortality in PAH, so being able to identify patients likely to respond to therapy, and conversely those who will not, carries important prognostic and treatment implications.17,74
Unfortunately, like other PAH directed therapies discussed above, relatively little is known about predictors of response to i.v. epoprostenol therapy prior to the initiation of the drug. In long-term follow-up studies of patients treated with i.v. epoprostenol, the strongest predictors of survival, like with other PAH trials and registries, are baseline clinical and hemodynamic characteristics indicative of severity of disease.17,74 McLaughlin et al. reported that change in PVR in response to adenosine challenge during initial RHC was predictive of improved survival with i.v. epoprostenol, but other studies using alternative means of acute VR are mixed in terms of prognostic value.17,44,45 Sitbon et al. examined clinical and hemodynamic parameters at baseline and after three months of therapy with i.v. epoprostenol, and found that decrease in total pulmonary resistance (TPR) by 30%, and increase in CI by 0.5 L/min/m2 at three months were associated with good response to therapy and long-term survival. 74 This is important because although it requires early and invasive hemodynamic assessment of response, being able to differentiate patients likely to do well with i.v. epoprostenol from those more likely to fail could help with deciding whom to refer for lung transplantation, which is likely the only viable treatment option for patients with progressive PAH despite i.v. epoprostenol.
“Super responders” to i.v. epoprostenol may not be a defined entity, but clearly represent a portion of the PAH population, with a subset of patients living over twice as long as would be predicted by recent registries, despite progressing to FC III–IV prior to the initiation of this therapy.
75
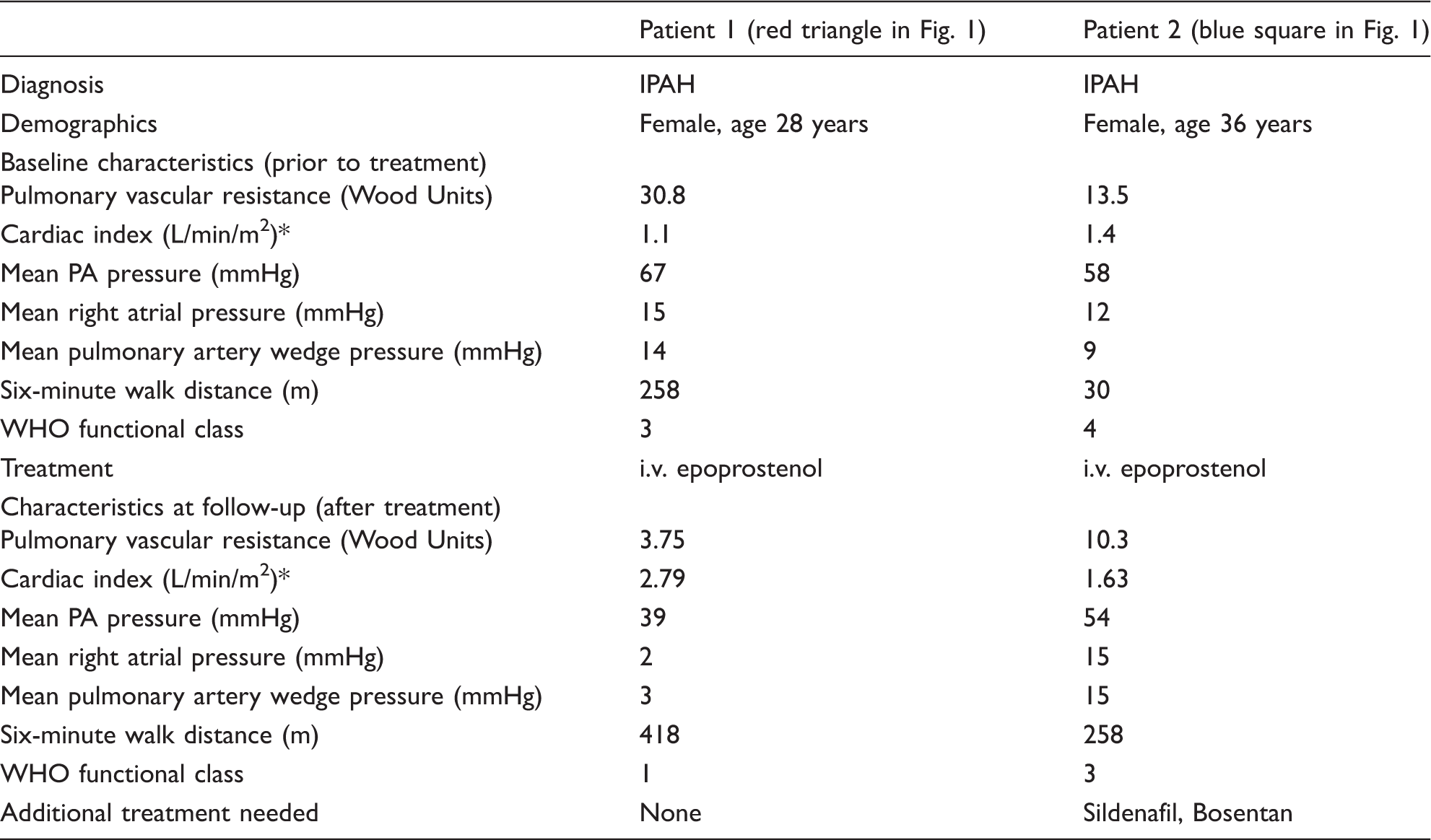
A representative example of the heterogeneity of response seen to i.v. epoprostenol is provided in Fig. 1, showing the change in PVR before and after the initiation of therapy, from a cohort of patients treated at our center. The clinical characteristics for two of these patients are provided in Table 2, highlighting the inter-individual variability in response to treatment, and the importance of personalized and precision medicine in PAH.
Change in PVR after initiation of i.v. Prostanoid. PVR in 27 patients with PAH with a RHC 17 ± 10 months after initiation of parenteral prostanoid therapy. Detailed data for two of the highlighted patients are provided in Table 2. Hemodynamic and clinical parameters of two IPAH patients treated with i.v. epoprostenol. Cardiac output measured by Fick’s method.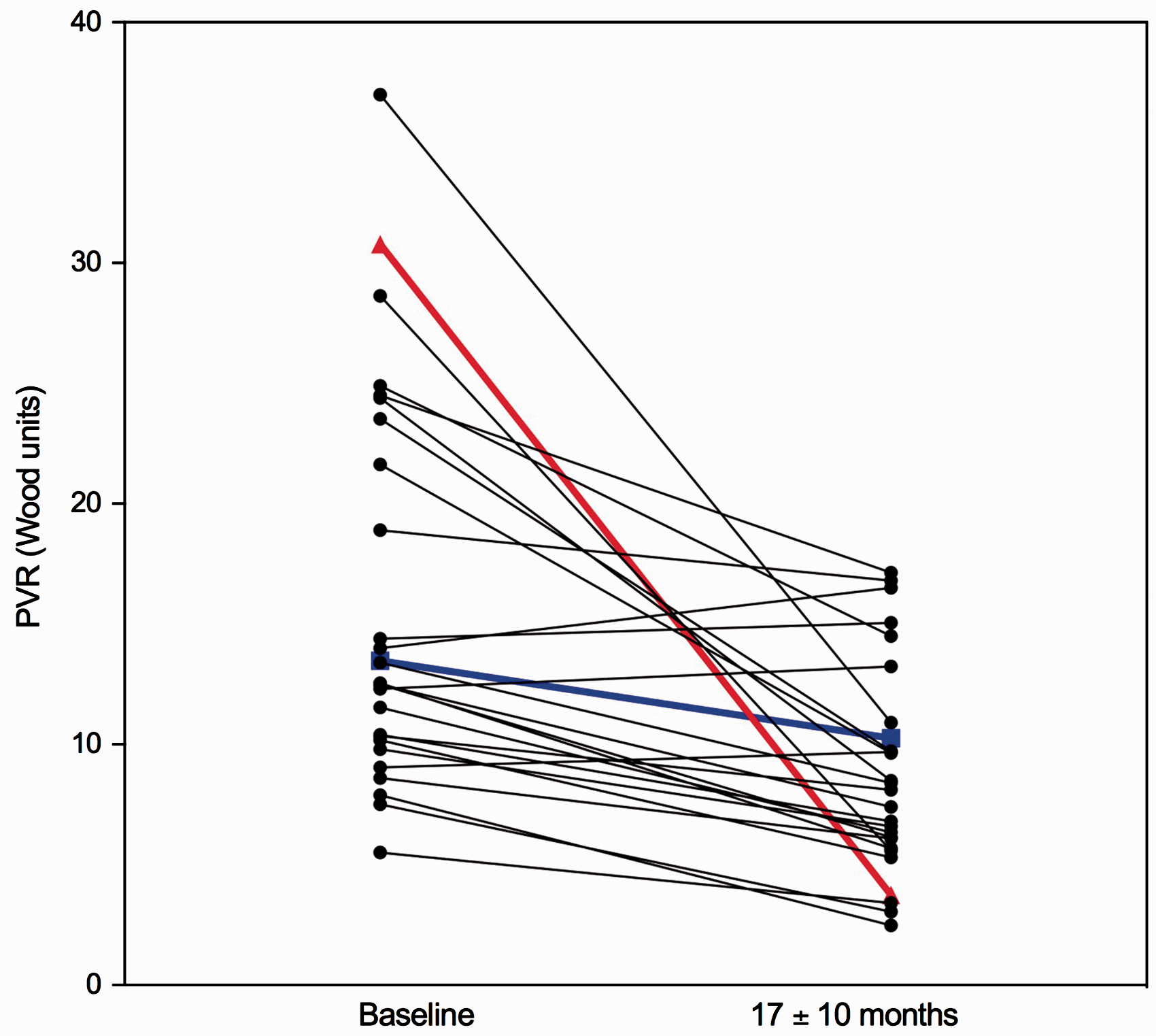
A case report of one such “super responder,” who survived for 18 years on i.v. epoprostenol without clinical progression before dying of colon cancer, provides interesting insights into what might characterize these patients and the pathogenesis of PAH. 75 Rich et al. found evidence of extensive proliferative vasculopathy and ongoing cellular proliferation in this patient at autopsy, but in contrast to patients who had died from PAH and RV failure, this patient’s RV showed evidence of hypertrophy and seemingly preserved contractility. 75 Preservation of RV function is crucial to survival in PAH, and presently little is known about what factors contribute to hypertrophy versus dilatation, but it appears to be a major determinate in the differential survival between men and women with the disease.1,76 There is emerging research that altered myocardial metabolism, specifically increases in glycolytic pathways and reduced fatty acid consumption, plays a major role in RV dysfunction via lipotoxicity, and further studies into this may allow us to better understand why some patients can have long-term survival on therapy despite persistently elevated PA pressures, while other have continued clinical deterioration.77–81
In addition to i.v. epoprostenol, there are two additional FDA-approved synthetic prostacyclin analogs, treprostinil, which is available in i.v., subcutaneous (SQ), inhaled, and oral (PO) formulations, and iloprost, which is available as an inhaled formulation.82–86 These all have the same mechanism of action, although the inhaled and PO formulations do not have the same long-term survival data as i.v. epoprostenol and are likely not as efficacious, but offer the considerable benefit of not requiring permanent i.v. access. Inhaled formulations have been used with success in patients who have complications or cannot tolerate parenteral prostacyclin analogs. 87 Being newer medications, there is less clinical and research experience with treprostinil and iloprost than epoprostenol, and it is not well-described if there are “super responders” to these therapies.
Prostacyclin receptor agonists
The newest approved medication for PAH is selexipag, an orally available selective IP receptor agonist. 88 The first of its class, selexipag is a structurally distinct molecule from prostacyclin analogs, but has similar mechanism of action and side effects.88,89 To date there has only been one large RCT of selexipag, which showed reduction in a composite end point of death or PAH complication (driven mostly by disease progression and hospitalization) in a cohort that included both treatment-naïve patients and patients on ERAs and PDE5 inhibitors. 88 With no published long-term follow-up data, and limited clinical experience with the drug, there are no known predictors of response at this time.
Combination and sequential therapies
With few treatment options, many patients who progress despite therapy, and proven efficacious drugs that work via different molecular pathways, it is intuitive that sequential and combination therapy has been a part of PAH therapy ever since there have been two drugs on the market. 90 The initial trial of upfront therapy with i.v. epoprostenol and PO bosentan did not show significant hemodynamic or clinical benefit, and due in part to negative early trials and in part to the high cost of PAH medications, a sequential combination approach had been favored over upfront combination therapy in both trials and clinical practice.2,90–94 Various sequential combination strategies of ERAs, PDE-5 inhibitors, and prostanoids have been used, with mixed success, but an overall trend has supported this strategy in patients who do not respond to initial monotherapy.2,95 There has been a recent change in this paradigm with the AMBITION study in 2015, in which patients treated with initial combination of ambristentan and tadalafil had improvement in a composite endpoint of death, hospitalization, disease progression, and clinical status compared to patients treated with monotherapy with either drug. 96 As a result of this trial, initial treatment with tadalafil and ambristentan now has a class I recommendation for WHO FC II and WHO FC III PAH patients in the most recent guidelines, though adoption of this strategy has been limited by the increased cost of starting two medications initially. Additionally, little is known about the characteristics of patients that may respond to initial combination therapy that would not have responded adequately to monotherapy, with fewer side effects and lesser cost. Perhaps some of the benefit seen in AMBITION can be attributed to patients with different likelihoods of deriving benefit from either an ERA or PDE5 inhibitor, as discussed in previous sections, having greater odds of receiving the right therapy upfront by receiving two drugs instead of one. As precision medicine in PAH advances, however, there will likely be a role for initial combination therapy in those patients with poor predictors of response to individual pharmacotherapies.
Summary
There have been tremendous advances in PAH therapy in the last two decades, with discovery and FDA approval of ten medications, in five classes, targeting three molecular pathways (Table 1). With the implementation of these medications into clinical practice, there has been an associated improvement in mean survival among PAH patients, from 2.8 years to greater than five years from time of diagnosis, albeit with potential confounding factors, such as lead time bias and selection of less ill patients for clinical trials.13,14 Despite these advances, however, PAH remains a progressive and ultimately fatal disease for most patients. With relatively few medications in development, improving outcomes for today’s PAH patients must focus on optimizing treatment for the individual using the available pharmacotherapy. Large RCTs have provided evidence that the available medications are safe and at least moderately efficacious across the PAH population as a whole, but PAH is a phenotypically and molecularly heterogeneous disease, so a more nuanced and personalized approach to therapy is needed to account for this inter-individual variability. One such way of doing this is by examining the patients who seem to have supra-normal response to the available therapies and learning from them so that future patients can hopefully derive similar benefits.
Identification of these “super responders” has long been recognized in pulmonary hypertension, dating back to the discovery of acute and long-term vasodilator responsive patients.9,40 An important lesson from the discovery of these 5–10% of IPAH patients who benefit tremendously from CCB therapy is that prior to refinement of acute vasodilator testing and the definition of vasodilator response, several vasodilators were tried for the treatment of PAH, often times with deleterious effect across the PAH patient population as a whole.39,41 This exemplifies the potential benefits of careful patient phenotyping and a shift from population wide to personalized medicine.
An interesting correlate to this experience is seen in the IMPRES trial of imatinib in PAH. 97 Imatinib, a tyrosine kinase inhibitor used for the treatment of chronic myeloid leukemia, had been shown in preclinical studies to have proapoptotic effects on pulmonary artery smooth muscle cells from IPAH patients, as well as vasodilatory effects in animal models of PAH.98–100 Spurred by these findings, several case reports emerged of PAH patients successfully treated with imatinib, with improvement in hemodynamics and functional class.101–103 A phase II study showed safety and tolerability of imatinib, and greatest improvement among patients with the highest PVR, which led to a phase III study of imatinib as an add-on therapy for patients with severe PAH.97,104 The IMPRES trial did, in fact, show that imatinib improved 6WMD and hemodynamics in patients with advanced PAH already on two to three PAH medications at the time of enrollment, some of whom improved dramatically.97,105 Enthusiasm for this finding was tempered, however, by the high rates of adverse events and drug intolerability, and the long-term extension was ultimately terminated early due to a high rate of adverse events, particularly subdural hematomas, and withdrawal from the study by both patients and the drug sponsor.97,106 Imatinib is not approved for treatment of PAH as a result of this trial, but those patients that responded well to treatment, perhaps representing a specific endophenotype of the disease, merit further investigation, and perhaps better characterization of this group could guide future research into which patients would be most likely to benefit from anti-proliferative chemotherapies. 107
Aside from the well-established acute vasodilator response criteria to detect “super responders” to CCB therapy, the ability to prospectively identify responders and non-responders to the currently available PAH pharmacotherapies is limited. Without a definition of “super responder,” the ability to study these patients and their unique characteristics is limited. There is some suggestion from retrospective analyses that men may derive more benefit from PDE5 inhibitors than women, and conversely, women may derive more benefit from ERAs.33,34 These results have not been verified prospectively, however, and in the limited number of head-to-head trials comparing ERAs and PDE5 inhibitors, there has not been a significant difference in primary outcomes between sexes.96,108 Presently, the only reliable clinical predictors of long-term response to medications in PAH remain baseline severity of disease and initial hemodynamic response to medications on follow-up evaluation. 74 However, with increasing availability of next generation sequencing and “omics” technologies, this paradigm is changing, and with incorporation of precision medicine into PAH, we may soon be targeting medications to “super responders.”
An early example of precision genomic medicine identifying “super responders” to medications in PAH can be seen in our group’s discovery of a gene expression signature that can be derived from peripheral blood and reliably distinguishes CCB responsive PAH from non-responders. 36 Benza and colleagues have also used genomic techniques to identify polymorphisms in the ET-1 pathway associated with positive response to ERAs. 35 Both of these studies require replication and prospective validation before clinical use, but gene-medicine interaction studies, such as these, could be the cornerstone of pharmacogenomics and precision medicine in PAH going forward. Our group is currently researching genetic associations with “super responders” to parenteral prostacyclin analogs. While exploring other clinical definitions of “super responders,” we have used the previously published short-term hemodynamic predictors of long-term survival after i.v. epoprostenol, as this definition offers one linked to survival. We encourage other groups to explore definitions of “super responders” to all classes of PAH medications and engage in a community-wide discussion of how to define these patients with the ultimate goal of generating a “super responder” definition used clinically and in practice, just as was done for CCB responders. 10
PAH patient registries have greatly increased our knowledge and understanding of the natural history of the disease in the past three decades, and combined with biobanking, will be key to better characterizing the endophenotypes of PAH to allow for precision medicine going forward. The first step is an agreed upon definition of “super responders,” which may rely on serial evaluations of clinical parameters such as 6MWT, laboratory parameters such an BNP, as well as invasive hemodynamic and echocardiographic measurements after initiation of pharmacotherapy. Identification of “super responders” will no doubt be difficult in the clinical trial setting, with increasing enrollment of older patients with multiple medical comorbidities, fewer FC III–IV patients included in trials, the high prevalence of patients on background therapy, and lack of long-term follow-up making true “super responders” harder to detect.109,110 However, most of the FDA-approved PAH medications have now been commercially available for a sufficient period to allow for retrospective analyses to see which of these parameters are associated with long-term response and survival. Once these “super responders” have been identified, via demonstration of improved clinical outcomes, the genomic, proteomic, and metabolomic data contained within the biobanks should be compared between these “super responders” and average or non-responders, both to allow for prospective identification of patients likely to respond to certain therapies and to enhance our understanding of the molecular mechanisms of the disease and drug response. Lastly, after development of these predictive “omics” profiles, prospective studies, stratified by “omics profile,” will be needed for confirmation of these findings.
Conclusions
PAH is a phenotypically and molecularly heterogeneous disease, and using the same medication algorithm for every patient, solely stratified by functional class and risk, will not be the treatment paradigm of the future, as our understanding of the multiple endophenotypes of the disease and pharmacogenetics advances. By careful phenotyping of patients, and better characterizing the molecular associations with response to pharmacotherapy, we will be able to tailor treatment to the individual, working toward a goal of making every PAH patient a “super responder.”
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.