
Abstract
Pulmonary arterial hypertension (PAH) is a devastating disease characterized by pulmonary vasoconstriction, pulmonary arterial remodeling, abnormal angiogenesis and impaired right ventricular function. Despite progress in pharmacological therapy, there is still no cure for PAH. The peptide apelin and the G-protein coupled apelin receptor (APLNR) are expressed in several tissues throughout the organism. Apelin is localized in vascular endothelial cells while the APLNR is localized in both endothelial and smooth muscle cells in vessels and in the heart. Apelin is regulated by hypoxia inducible factor-1α and bone morphogenetic protein receptor-2. Patients with PAH have lower levels of plasma-apelin, and decreased apelin expression in pulmonary endothelial cells. Apelin has therefore been proposed as a potential biomarker for PAH. Furthermore, apelin plays a role in angiogenesis and regulates endothelial and smooth muscle cell apoptosis and proliferation complementary and opposite to vascular endothelial growth factor. In the systemic circulation, apelin modulates endothelial nitric oxide synthase (eNOS) expression, induces eNOS-dependent vasodilatation, counteracts angiotensin-II mediated vasoconstriction, and has positive inotropic and cardioprotective effects. Apelin attenuates vasoconstriction in isolated rat pulmonary arteries, and chronic treatment with apelin attenuates the development of pulmonary hypertension in animal models. The existing literature thus renders APLNR an interesting potential new therapeutic target for PH.
INTRODUCTION
Pulmonary hypertension
Pulmonary arterial hypertension (PAH) is a severe disease with a median survival of 2.8 years if left untreated.[1] Over the past two decades, novel drugs with a pulmonary vasodilator action and a possible additional inhibitory effect on vascular cell proliferation have been developed, but even after the introduction of such compounds the chance of survival remains poor, with a 3-year survival less than 60%.[2]
PAH is characterized by a mean pulmonary arterial pressure (MPAP) above 25 mmHg at rest and an increased pulmonary vascular resistance (PVR) in combination with a normal pulmonary capillary wedge pressure (PCWP).[3] PAH eventually leads to right ventricular pressure overload and compensatory hypertrophy followed by dilatation and failure of the right ventricle,[4,5] which is the most common cause of death.[6] The current therapeutic drugs are primarily pulmonary vasodilators such as endothelin-1 (ET-1) receptor antagonists, prostacycline analogues and phosphodiesterase-5 inhibitors that aim to correct for abnormalities in the secretion of endothelium-derived vasoactive mediators. Nevertheless, no current therapy against PAH is sufficient to cure or stop the disease progression. Consequently, there is a need for new therapies.
Pathophysiological mechanisms of PAH
Multiple genetic, cellular and molecular functions are involved in the pathophysiology of PAH. These have recently been reviewed extensively.[7] A number of pathophysiological mechanisms involved in PAH are relevant in relation to the subject of this paper. For example, normoxic activation of hypoxia-inducible factor (HIF-1α), normally exerting the physiologic hypoxic vasoconstriction, can occur in cells prior to the spontaneous development of PAH in fawn-hooded rats and is thought to be a possible contributor to the development of PAH.[8] Furthermore, genetic aspects play a role. One of the most prominent genes involved in PAH is the bone morphogenetic protein receptor 2 (BMPR-2), in which mutations occur in 70% of patients with familial PAH and in 25% of patients with idiopathic PAH.[7]
Abnormal apoptosis and proliferation of vascular endothelial and smooth muscle cells,[7,9] is involved in the remodeling process of the pulmonary arteries, development of plexiform lesions, and loss of the microvasculature. Numerous humoral factors, including vascular endothelial growth factor (VEGF), are involved in this response.[9] Furthermore, the function of the endothelium is altered in PAH, resulting in an imbalance between endothelium-derived vasoconstrictors and proliferative agents such as ET-1 and thromboxane, and vasodilators with antiproliferative effects including nitric oxide (NO) and prostacyclin.[10] In addition to contributing to the remodeling process, it results in decreased vasorelaxation of the pulmonary vascular bed. Angiotensin-II also induces vasoconstriction and mitogenesis in PAH, while enhanced expression of the angiotensin-II converting enzyme 2 (ACE2) has been found to have a beneficial effect in animal models of pulmonary hypertension.[11,12]
The right ventricle is subjected to pressure-induced alterations in PAH. Compensatory hypertrophy and fibrosis of the right ventricle develops, followed by decreased systolic function and dilatation.[4] Among other mechanisms, ischemia and apoptosis are central players in this process,[4] and have increased the interest to investigate whether drugs directly targeting mechanisms in the right ventricle may improve the course of PAH.
Apelin and the apelin receptor
The peptide apelin and the apelin receptor (APLNR) are present in the heart,[13,14] the systemic and pulmonary vasculature, and the expression of apelin and APLNR is regulated by HIF-1α[15] and BMPR-2.[16] Furthermore, the apelin-APLNR system is involved in normal vascular development[17] and regulation of apoptosis,[16] and has been shown to be involved in regulation of NO-dependent vasodilatation[15,18] and to improve cardiac contractility.[19] Most studies on apelin have addressed the systemic circulation, but the characteristics revealed by these studies have drawn attention to apelin-APLNR as a potential new target in pulmonary hypertension. Recently, support for a role of apelin and APNLR in pulmonary hypertension has emerged through experiments in animal models of pulmonary hypertension.
The purpose of this paper is to introduce the apelin-APLNR system, to review the associations between PAH pathophysiology and apelin based on existing studies, to touch upon apelin as a potential biomarker for PAH, and to point out the regulating effects of apelin on vascular endothelial and smooth muscle cell proliferation, the vasodilatory effect and positive inotropic effect that renders modulation of APLNR an interesting therapeutic target in PAH.
DISCUSSION
Apelin and APLNR
Structure
APLNR was discovered in 1993 by O'Dowd and colleagues who were looking for subtypes of the vasopressin receptor The APLNR was first named APJ and is a G-protein coupled 7 trans-membrane receptor coded for on chromosome 11 band 12, and shares 54% homology in the trans-membrane region with the AT1 receptor. It does not, however, bind angiotensin-II.[20] Human APLNR shares 74% homology with the rat APLNR.
In 1998, apelin was isolated by Tatemoto et al.[21] They showed that the active forms of apelin are peptides cleaved from the C-terminal of the 77 amino acids long preproapelin. Fragments of 36, 17, 13 including the N-terminal pyroglutamated apelin-13 (Pyr1-apelin-13), exist in vivo and have biological activity, but also synthetic fragments of the 12 amino acids from the C-terminal can activate the receptor.[22] It has been suggested that PyrJ-apelin-13 is the final active product of apelin, which is more resistant to enzymatic cleavage.[23,24] Biological differences between apelin fragments have been shown[25] (e.g., apelin-13, and −17 binds the receptor with higher potency), whereas it is proposed that apelin-36 dissociates slower from the APLNR.[26–28]
ACE 2 cleaves a single amino acid from the c-terminal of apelin-13 by hydrolysis[29] (Fig. 1a). It is not fully elucidated whether this inactivates the protein, since studies investigating apelin-17 with amino-acids deleted from the c-terminal found that such peptides still bound to the receptor.[30] However, the study suggested that receptor activation was altered, in that internalization did not take place.[19] So far, ACE 2 is the only known enzyme metabolizing apelin. Nevertheless, it seems that the half-life of apelin is short. A half time less than 8 minutes has been documented in humans.[27]
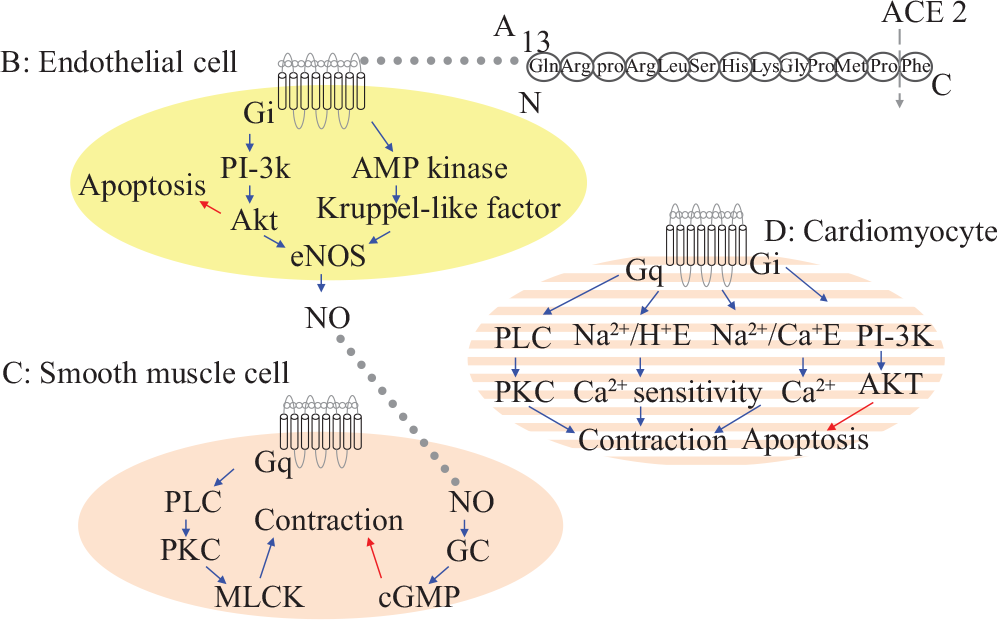
Proposed signaling pathways for apelin and APLNR. The figure summarizes data obtained from both systemic and pulmonary vasculature and both left and right ventriclet. A: the structure of apelin-13 showing the site for ACE-2 mediated hydrolysis. B: known pathways in endothelial cells, C: vascular smooth muscle cells, and D: cardiomytes, blue →: Stimulates activity; red →: Decreases activity. Na2+/Ca2+ E: Na2+/Ca2+ exchanger, Na2+/H+ E: Na2+/Ca2+ exchanger.
In short, the difference between the apelin-peptides of different lengths is not fully determined, and furthermore, the pharmacokinetics of the peptides are largely unknown.
Apelin-APLNR interaction and signaling
There is evidence that the APLNR is coupled to an inhibitory Gi-protein because apelin inhibits forskolin induced 3′5″ cyclic adenosine monophospate (cAMP) production in Chinese hamster ovary (CHO) cells[21,26,31] and because extracellular acidification in CHO cells can be blocked by pertussis toxin.[26] Furthermore, apelin also works by stimulating the Na2+/H+ exchanger in CHO cells,[26] and apelin has been shown to activate extracellular signal-regulated kinase (ERK).[28,32] Some effects of apelin have been shown to be mediated by the phophoinositol-3-phosphokinase (PI-3K)/AKT activated pathway.[33] Although Masri et al. reported that apelin could not induce increments of inositol phosphate in CHO cells and concluded that the APLNR did not bind a Gq/G11 protein,[32] the positive inotropic effects of apelin are dependent on the function of phospholipase C (PLC), protein kinase C (PKC), the Na2+/H+ and the Na2+/Ca2+ exchanger,[19] and an increase in intracellular Ca2+ transients.[34] These findings are consistent with coupling of the APLNR to a Gq-protein[35] (Fig. 1).
A 2004 study found that the APLNR was internalized into the cytoplasm and subsequently, the nucleus, upon apelin-APLNR interaction in CHO cells, and it was observed that only apelin fragments able to induce internalization exerted a biological in vivo response.[30] However, other G-protein coupled receptors such as the angiotensin-II receptor AT1 are able to elicit biological responses both through activation of the G-protein, but also to act on intracellular pathways after internalization.[36] Hence, this coupling between internalization and effect requires further investigation.
To date, apelin has not been proven to bind to other receptors,[31,37,38] and no subtypes of the APLNR have been found. Also, no other endogenous ligands for the APLNR are known, but mice lacking the APLNR are different, as described below, from mice lacking the apelin gene, which suggests that other endogenous substances influence the activity of APLNR.[39] For example, the APLNR has been shown to form heterodimers and oppose the effect of the angiotensin-II receptor independently of the presence of apelin.[40]
Localization of APLNR and apelin
Early studies showed that apelin and APLNR mRNA was expressed in numerous tissues of the rat,[26,41] including several areas of the brain, adipose tissue, heart and lungs. The expression in the lungs was greater than in other tissues, except for a very high expression of apelin in the mammary gland.[41] In the cardiovascular system, APLNR immunoreactivity was present in both endothelial and smooth muscle cells in the vasculature, in endocardium and myocardium. Apelin immunoreactivity was present in the myocardium, endocardium, and in endothelial cells of the large conduit vessels. In the lungs, apelin immunoreactivity was demonstrated only in the endothelium of small pulmonary arteries, not in the epithelium or vascular smooth muscle cells.[14] Apelin immunostaining was localized to vesicle-like structures in the endocardial endothelium and in structures associated to the nuclear surface in endothelial cells.[13] The APLNR has been demonstrated in nuclear fragments of cells.[42] On the basis of these observations, it has been suggested that apelin is secreted by endothelial cells[13] and that the APLNR may affect regulation of gene transcription.[42]
In human tissue, the relative expression of apelin differs from the rat as the mRNA expression of preproapelin and the apelin receptor in the spleen and central nervous system seems higher than in the lungs.[43] Apelin was also shown to be present in the endothelium of small and larger pulmonary vessels in human tissue.[44]
Accordingly, the location of apelin in the pulmonary endothelium with receptors in both endothelium and smooth muscle makes this system interesting in relation to pulmonary vascular disease.
Regulation of apelin and APLNR
Factors regulating apelin and APLNR are summarized in Figure 2.
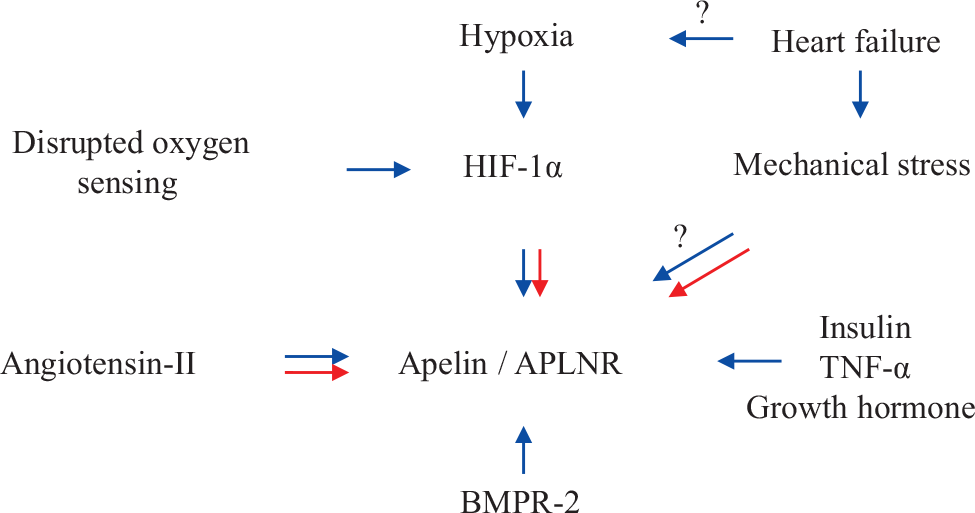
Factors influencing expression of apelin. Blue →: Stimulates expression; Red →: Decreases expression.
Hypoxia
Vast evidence supports the assertion that apelin expression is regulated by hypoxia. In mice, an increase in total pulmonary apelin mRNA was reported to take place after 1 week of hypoxia,[45] and in another study in mice, approximately a doubling of apelin mRNA and an increase of apelin protein of about 25% was found after 5 hours of hypoxia.[46] In rats, however, total pulmonary apelin content was unchanged after exposure to hypoxia for 2 weeks.[47] An explanation of this apparent incongruence was demonstrated in cultured pulmonary endothelial cells, where apelin mRNA levels were up-regulated after 8 hours of hypoxia, but fell after further hypoxic exposure.[46] In a study made on mice by Chandra et al., a similar tendency was shown in right ventricular tissue. Here, apelin and APLNR mRNA was four- and three fold increased after one week of hypoxia, but unchanged after 3 weeks.[48] An initial up-regulation followed by a decrease is also found after glucose deprivation in cultured cardiomyocytes.[33] The hypoxia-induced regulation of apelin and the APLNR was also found in cultured human hepatocytes, stellate cells[49] and adipocytes,[15,50] where APLNR and apelin were up-regulated within 24 hours of hypoxic exposure. Similarly, secretion of apelin to extracellular medium is increased by short-time hypoxia in cultured cardiac myocytes[51] and adipocytes.[50] The regulation induced by hypoxia was shown to be mediated by HIF-1α in adipocytes.[15,50]
In short, it seems that hypoxia regulates the apelin pathway in various tissues, and that there is a biphasic response of apelin and the APLNR to hypoxia with an initial up-regulation followed by a normalization or decrease. Interestingly, in the context of pulmonary hypertension, HIF-1α is a mediator for the hypoxia induced regulation of apelin and APLNR.
Heart failure—mechanical strain or hypoxia?
Apelin and APLNR are altered in heart failure. In an animal model of ischemic heart failure, total heart apelin protein was up-regulated 6 weeks after induction of myocardial infarction.[52] A study in Dahl salt-sensitive hypertensive rats showed that apelin and APLNR mRNA expression was unaltered in a phase of compensated left heart hypertrophy, but decreased in manifest heart failure,[53] and in a model of isoprenaline-induced heart failure, apelin and APLNR were found to be down-regulated.[54]
In one human study, APLNR mRNA and apelin protein levels were up-regulated in left ventricular tissue, but lower in atrial tissue from patients with heart failure compared to healthy controls.[55] Other studies addressing plasma levels of apelin in heart failure patients have reported down regulation of the apelin system.[44,56,57] In line with the proposed biphasic response to hypoxia, it is also possible that apelin and APLNR are regulated in opposed directions during different phases of heart failure. This is supported by a study by Chen et al., who showed that apelin plasma levels were in fact higher in patients with low NYHA class heart failure than in healthy controls, but that the levels declined in more advanced functional classes.[58] Interestingly, cardiac APLNR mRNA rose in patients after implementation of a left ventricular assist device.[58] This could point towards a regulatory effect of mechanical strain on the heart or other factors involved in heart failure e.g., humoral mediators. To support this, apelin and APLNR were down-regulated in the right ventricle of monocrotaline rats with pulmonary hypertension[23] and in mice subjected to aortic banding.[59] On the other hand, tissue hypoxia may also be present in these types of heart failure, and the hypoxia stimulus attenuated by improving left ventricular function. This topic therefore needs more investigation. Apelin levels were also shown to be decreased in patients with ischemic heart disease,[60–62] and again both hypoxia and mechanical stress may be involved.
BMPR-2, angiotensin-II, insulin and other regulatory factors
A link between the BMPR-2 and apelin was recently recognized.[16] It was shown that disrupted BMPR2 signaling mediated through Peroxisome proliferator-activated receptor gamma (PPARy)/β-catenin resulted in decreased apelin expression and increased endothelial cell apoptosis. In addition, the authors showed that apelin secreted from pulmonary endothelial cells inhibited proliferation of pulmonary arterial smooth muscle cells. The authors suggested that apelin is a downstream protein from the BMPR-2 receptor signaling involved in pulmonary vascular homeostasis.
Treatment with angiotensin-II has been shown to up-regulate APLNR expression in cultured hepatocytes.[49] In models of decompensated heart failure APLNR expression was decreased,[53,63] and in one study, this was prevented by treatment with an angiotensin-II receptor antagonist.[53] A β-adrenoceptor agonist failed to do so, even though the two drugs improved the systolic function of the left ventricle equally.[53] These results are apparently contradictory, but indicate a regulatory relationship between the apelin and angiotensin-II systems.
In adipose tissue, insulin up-regulates the expression of apelin in obese mice and insulin levels are related to plasma apelin levels in obese individuals.[64] Furthermore, growth hormone and tumor necrosis factor-α are proposed as regulators of apelin in adipose tissue.[65,66]
Overlap between apelin regulation and factors involved in PAH
Accordingly, apelin and APLNR are regulated by hypoxia, which is an important factor in pulmonary vasoconstriction. In addition, HIF-1α, BMPR-2 and angiotensin-II that have all been linked to the pathophysiology of pulmonary hypertension also regulate apelin/APLNR expression. Furthermore, it is possible that mechanical strain and cardiac overload, that are also features of pulmonary hypertension, affects apelin-APLNR levels. Two studies have shown that serum apelin levels in patients with PAH are lower than in controls,[44,48] and also a decreased content of apelin in pulmonary arterial endothelial cells in patients with PAH has been demonstrated.[16] This supports a role of apelin and APLNR in pulmonary hypertension. The regulation by insulin makes it interesting to investigate the role of apelin in obesity-related pulmonary hypertension.
Apelin as a plasma biomarker
The changes in plasma apelin levels in heart failure[44,55–58] and pulmonary hypertension[44,48] have brought forward the idea of apelin as a possible biomarker for these diseases. In heart failure patients,[55] the concentration of apelin was 200 fold higher in atrial tissue than in left ventricular tissue. Plasma apelin concentrations correlated to atrial apelin levels, and it was therefore suggested that atrial apelin might be an important source of apelin in plasma.[55]
However, studies have compared apelin with brain natriuretic peptide (BNP) and the split product N-terminal BNP (NT-proBNP) as biomarkers in heart failure. It has been established that the main source of circulating BNP and NT-proBNP is the heart,[67] and in patients with myocardial infarct, apelin-36 was inferior to BNP in order to detect decreased left ventricular systolic function.[68] Similarly, other studies report that NT-proBNP is superior to apelin in correlation with heart failure severity[69] and prediction of mortality in acute heart failure.[70]
In pulmonary hypertension, apelin was proposed as a potential lung-derived biomarker, because of the high expression of apelin in the lungs.[44] If pulmonary apelin was reflected in plasma, it would be a very interesting biomarker yielding information about the small pulmonary artery endothelium given the presence of apelin here.[13] Nevertheless, an animal study in chronic hypoxic rats with pulmonary hypertension showed that apelin content in the lungs were not reflected in plasma samples and that plasma apelin levels did not correlate to right ventricular pressure[47] (Fig. 3). On the other hand plasma apelin levels correlated weakly (r2 = 0.33) to changes in right ventricular apelin levels (Fig. 3).
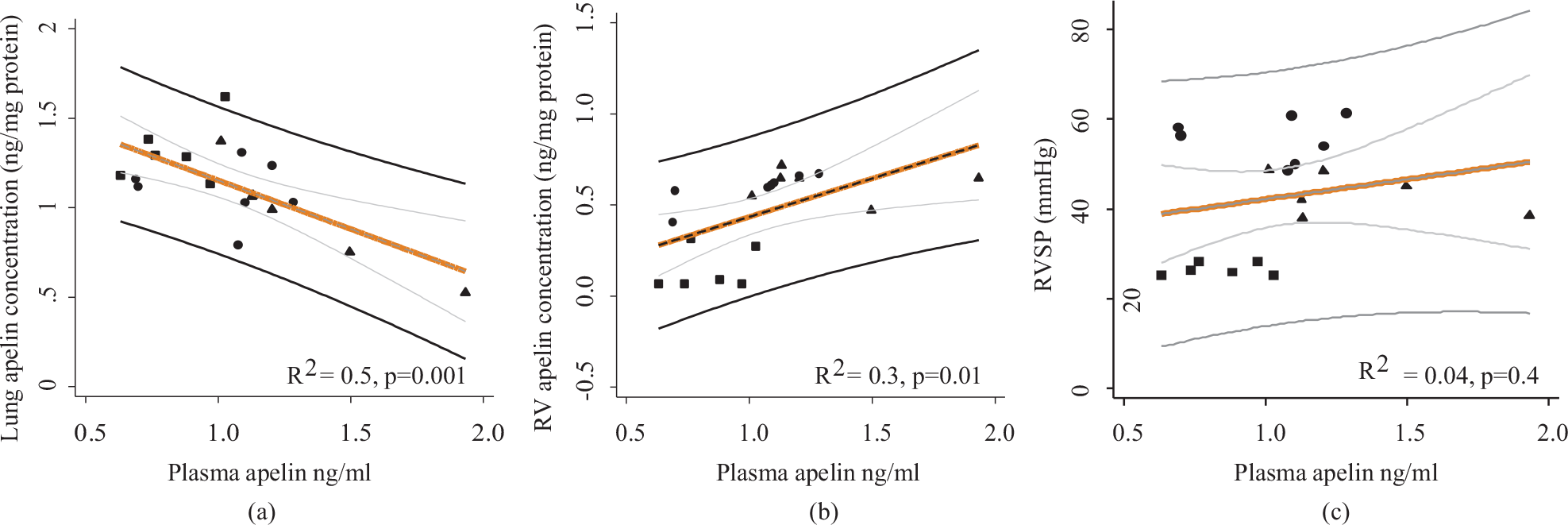
Original figure adapted from data in Andersen et al.[47] (a): In chronic hypoxic rats, lung concentrations of apelin were not altered in the same direction as plasma apelin by hypoxia and sildenafil treatment. (b): Right ventricular apelin concentrations correlated weakly with plasma apelin levels; (c): Plasma apelin levels were not correlated to right ventricular systolic pressure. RVSP: Right ventricular systolic pressure. ■ = Normoxic rats; ● = Hypoxic rats, ▲ = Hypoxic rats treated with sildenafil 25 mg/kg/day.
In short, it is not fully established, which organs are most important in determining the plasma levels of apelin, but the widespread distribution of apelin probably reduces the use of the peptide as a marker for functional change in a specific organ. At least apelin does not seem to be a lung-derived biomarker for pulmonary hypertension, and, extrapolating the results from studies in patients with left heart disease, the BNPs may be expected to be more specific than apelin in detecting right ventricular failure secondary to pulmonary hypertension.
Effect of apelin in angiogenesis and vascular cell homeostasis
Angiogenesis
In 1996 the APLNR was found to be present in endothelial cells and precursor cells of the embryonic vasculature.[17,71] Disrupting the apelin/APJ pathway resulted in reduction or loss of intersegmental vessels in frog embryos,[72] and the authors also showed that apelin was a mitogen and migration factor for endothelial cells. Mice lacking the apelin gene are viable, and postmortem analyses have shown no major anatomical or histological abnormalities. However, APLNR deficient mice show abnormalities in regulation of blood pressure, and in the cardiovascular development. Furthermore, the offspring of APLNR heterozygous mice did not follow a Mendelian order, showing a significantly smaller number of APLNR−/− mice than expected.[39] This suggests that the APLNR plays a more pivotal role than apelin per se, and as mentioned previously, that the APLNR may be activated by other ligands.
Apelin has also been shown to be an angiogenic factor in retinal cells.[73–77] Lack of apelin has been shown to decrease the hypoxia-induced regeneration of vessels in zebra fish,[46] and in accordance, a study from 2010 showed that lack of apelin worsens necrosis in a model of hind limb ischemia in mice. Furthermore, apelin administration was able to increase the number of larger vessels and decrease necrosis in a model of hind limb ischemia in mice.[78]
Apelin and VEGF—similarities and differences
Like vascular endothelial growth factor (VEGF), apelin inhibits apoptosis in endothelial cells.[16,79] There are conflicting results about the effect of apelin on apoptosis and proliferation in vascular smooth muscle cells. Some studies found that apelin inhibits apoptosis in umbilical vein smooth muscle cells,[80] and stimulates proliferation in vascular smooth muscle cells from rat thoracic aortas[81,82] evoked by activation of the PI3-K/AKT pathway.[80,81,83] However, a study investigating the effects in pulmonary artery smooth muscle cells found that apoptosis was increased by apelin.[16] Another characteristic of apelin similar to VEGF is a stimulatory effect on tumour and micro-vessel growth in relation to cancer.[80,83,84]
In contrast to apelin or APLNR deficient mice, homozygous VEGF knockout mice die mid-gestational due to severe developmental abnormalities of the circulatory system.[85] This suggests that VEGF is more important in vascular development than apelin. The case seems to be an inter-relationship between apelin and VEGF in that apelin stimulated the expression of VEGF-A in cell culture.[49] Moreover, VEGF and apelin combined has been shown to more effectively establish new vessels in animal models of limb ischemia;[78] and, interestingly, apelin counteracted the increased vascular permeability of new vessels induced by VEGF.[78] Increased vascular leakage may be involved in the pathobiology of PAH,[7] and apelin has been shown to reduce lipopolysaccharide-induced vascular leakage in rat lung tissue.[86]
PAH, apelin and VEGF
In PAH patients, endothelial cells have been shown to be abnormal in that proliferation and migration is enhanced, while angiogenesis is less effective in order to generate structured networks,[87] and other studies also point toward a disordered angiogenesis in PAH.[88]
The role of VEGF in pulmonary hypertension is ambiguous since the lack of VEGF clearly worsens the severity of pulmonary hypertension development in animal models,[89] due to apoptosis of normal endothelial cells. This leads to recruitment of poorly differentiated apoptosis-resistant endothelial cells forming plexiform lesions,[90–92] and furthermore, smooth muscle cells proliferate abnormally.[16] On the other hand, it seems that excessive VEGF in advanced stages of PAH contributes to the abnormal endothelial cell proliferation and generation of plexiform lesions.[93] So far apelin seems to be favorable in pulmonary hypertension. The study by Chandra et al. showed more severe pulmonary hypertension and a more pronounced loss of microvessels in the pulmonary vasculature in chronic hypoxic mice lacking the apelin gene.[48] This, taken together with the attenuating effect of apelin on pulmonary endothelial cell apoptosis,[16] may suggest that apelin could modulate the angiogenesis in a positive way in PAH. In pulmonary hypertension related to scleroderma and pulmonary fibrosis, a decreased vascular capacitance is of particular importance,[94,95] and the effects of apelin in these diseases are worthy of further elucidation.
Effects of apelin on vascular tone and blood pressure
Isolated arteries
In isolated systemic artery[96,24] and vein[97] preparations, apelin reduces vascular tone by 25%-50%, and vasoconstriction in response to angiotensin-II has been shown to be inhibited by incubation of arteries with apelin.[98,99] Other studies[100] confirm the modulatory effect of apelin on angiotension-II induced vasoconstriction, including a study in APLNR deficient mice that had increased pressor response to angiotensin-II.[38] However, it was observed that apelin exerted vasoconstriction in denuded vena saphena magna preparations.[37] In keeping with this, apelin phosphorylates myosin light chain kinase dependent on PKC in vascular smooth muscle.[101] This suggests that the apelin induced vasodilatation is endothelium-dependent, and that apelin has the opposite effect when acting directly on the smooth muscle cells (Fig. 1).
Blood pressure
One of the first proven physiological effects of apelin was the ability to temporarily lower blood pressure after injection in rats,[18] and several studies have confirmed this effect,[24,25,30,38,102] including studies showing vasodilation in human volunteers and heart failure patients[27, 103] (Table 1). However, a single study found that apelin given intravenously resulted in an increment of blood pressure in conscious rats[105] (Table 1). Furthermore, apelin injected or overexpressed in the rostral ventrolateral medulla of the central nervous system results in blood pressure increments.[106,107]
Effects of apelin on blood pressure in the systemic circulation
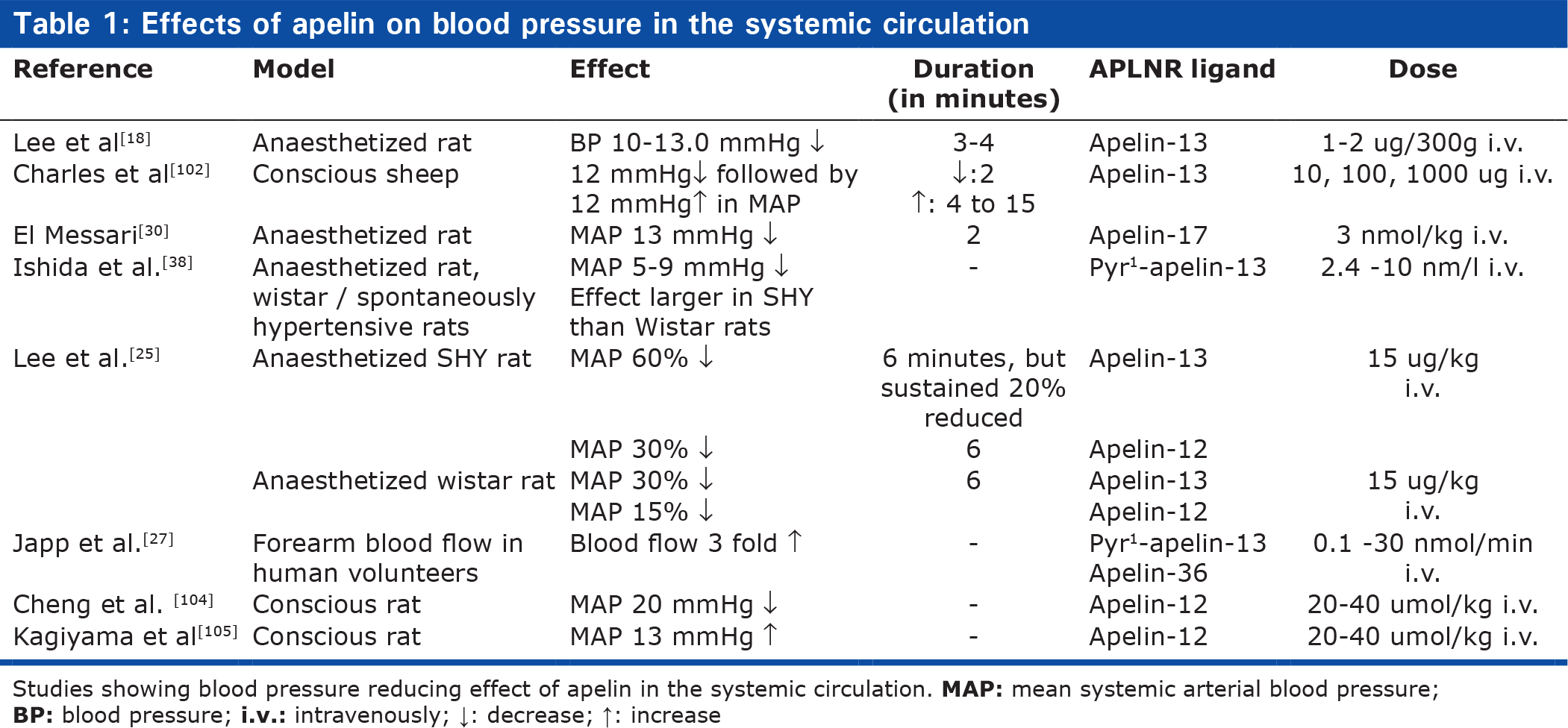
Studies showing blood pressure reducing effect of apelin in the systemic circulation.
The results suggest that the effects of apelin on blood pressure is not straightforward, and that the net result may depend on the apelin fragment, way of administration, conscious state of the experimental animal, and the propensity for compensatory increases in heart rate and cardiac output.
Apelin and the endothelial NO synthase
Inhibition of prostaglandin synthesis has been shown to both reduce[24] and be without influence[97] on the effects of apelin, while numerous studies show that the vasodilatory effect of apelin is inhibited by inhibition of endothelial NO synthase (eNOS).[16,27,38,96,97]
It is also evident that apelin stimulates the activity of eNOS.[38,98,99] Apelin near-normalized the eNOS phosphorylation in in vitro preparations of the aorta[99] and renal artery[98] in diabetic mice with endothelial dysfunction. On the contrary, apelin-induced vasodilatation was reduced in hepatic arteries from patients with liver cirrhosis compared to mesenteric arteries from healthy donor patients,[96] and this was suggested to be due to a decreased function of eNOS in the arteries from liver cirrhotic patients. However, caution should be taken interpreting these data, since two different vessel types are compared. In short, it is controversial whether apelin normalises impaired eNOS function and improves endothelial function in disease states, or if apelin looses its effects in case of endothelial dysfunction because of decreased eNOS function. AKT,[99] AMP kinase[48] and kruppel-like factor 2[48] have been suggested to link stimulation of APLNR to eNOS (Fig. 1).
Effect of apelin on pulmonary arteries and pulmonary pressure in vitro and in vivo
The effect of apelin in pulmonary arteries has been examined in only a few in vitro studies.[47,108] One group found that only very high concentrations of apelin were able to transiently decrease the vascular tone with maximally 17%. In a Chinese study, apelin decreased tone by maximally 11%, and the vasorelaxation was abolished by removal of the endothelium and treatment with the eNOS blocker L-NAME[108] in line with the findings in the systemic circulation. Furthermore, the Chinese group found that the relaxation was decreased by 60% in isolated pulmonary arteries from hypoxic animals. Accordingly, it was shown that in isolated pulmonary resistance arteries from normoxic control rats, treatment with apelin inhibited vasoconstriction to endothelin-1,[47] while this effect was absent in hypoxic rats with pulmonary hypertension. There was no down-regulation of the APLNR in the arteries from hypoxic rats,[47] suggesting that the impairment was downstream to the apelin receptor in pulmonary hypertension. This favors the hypothesis that vasodilation induced by apelin is decreased in disease states.
In vivo experiments with dogs subjected to acute pulmonary embolism, showed that apelin injected as a single dose of 20 mg/kg slightly reduced mean pulmonary arterial pressure for approximately 2 minutes. However, the ratio of pulmonary to systemic vascular resistance was not changed.[109] Long-term studies of apelin in animal models of pulmonary hypertension have shown that it has an attenuating effect on the pulmonary pressure.[16,23,110] It is, though, difficult to say if this is due to direct vasodilatation, to counteraction or down-regulation of constrictive mediators,[23] to decreased loss of endothelial cells[16] and microvasculature,[48] or to inhibition of smooth muscle cell proliferation.[16]
PAH, angiotensin-II and apelin
Angiotensin-II has been proposed to contribute to the increased pressure and remodeling in PAH,[12] and an attenuating effect of apelin on angiotensin-II mediated vasoconstriction in isolated pulmonary arteries from normoxic rats has been observed[47] in accordance with findings in the systemic circulation. In hypoxic rats with pulmonary hypertension, the inhibitory effect was not present, but this does not exclude that long-term apelin treatment could modulate the effect of angiotensin-II. For example, in cardiomyocytes, apelin blocked angiotensin-II-induced activation of the Rho-A kinase pathway,[100] which plays a pivotal role in pulmonary hypertension;[7] and, furthermore, apelin has been shown to antagonize angiotensin-II-induced gene transcription.[40] The significance of apelin and angiotensin-II sharing ACE 2 as a part of their degradation pathway remains enigmatic in relation to pulmonary hypertension. Exogenous activation of ACE 2 with a synthetic activator has been shown to prevent monocrotaline-induced pulmonary hypertension in rats,[111] because of an increased breakdown of angiotensin-II to the vasodilatory angiotensin(1-7). How this beneficial effect agrees with increased hydrolysis of apelin by ACE 2 is yet unexplored.
Cardiac effects of apelin-APLNR
Effect on cardiac contractility
An important role for apelin in cardiac function was shown by results from Kuba et al. who observed that apelin-deficient mice developed impaired cardiac contractility with age, unlike their wild type littermates.[59] Apelin has also been proven to be a potent positive inotropic agent. This was shown for the first time by Szokodi et al. using an isolated perfused heart preparation.[19] This study showed that apelin was able to increase contractility (by 60% of developed tension) independently of angiotensin-II receptors, endothelin-1 receptors or α-and β-adrenoceptors, and independently of eNOS.
Furthermore, they showed that the positive inotropic effect was reduced by the inhibition of PLC and PKC as well as the Na2+-H+ exchanger and Na2+-Ca2+ exchanger (Fig. 1). In this study, very low concentrations of apelin-16 (0.01-10 nM) were used, and thus apelin was characterized as one of the most potent positive inotropic agents known. The inotropic effect has been confirmed by other studies,[34,52,54,112] and interestingly, in normal wild type mice, two weeks infusion of apelin resulted in increased cardiac output, but with no evidence of cardiac hypertrophy.[113]An interesting report from Japp et al showed increased coronary blood flow, decreased blood pressure, increased contractility and increased cardiac output following acute apelin administration in patients with heart failure.[103] Some groups have found that the increased contractility was associated with an increment of intracellular Ca2+,[34] while other studies have shown no increases in Ca2+ transients, but a possible higher myofilament sensitivity towards Ca2+.[114]
Cardioprotective effect of apelin
Improved systolic cardiac function[115] and reduction in infarct size[115,116] have been observed in isolated rat hearts after ischemic insult and reperfusion. This was associated to decreased apoptosis,[33,115] and in the majority of studies,[33,115,117] the PI3/AKT pathway has been found to mediate this effect (Fig. 1). Another cardioprotective role of apelin is the ability to reduce angiotensin-II-induced cardiac fibrosis.[100]
Right ventricle, apelin and PAH
The findings of improved contractility without concomitant hypertrophy, and cardioprotective abilities are intriguing in the setting of pulmonary hypertension, because the final consequence of the disease is right ventricular hypertrophy, fibrotic remodeling, and failure followed by death. It has been shown that apelin increases contractility in failing right ventricular myocardium from animals with pulmonary hypertension.[34] A study addressing the effect of apelin in monocrotaline induced pulmonary hypertension found that apelin was able to normalize contractility indices (dP/dtmax and dP/dtmin) of the right ventricle measured in vivo compared to control animals.[23] However, it is uncertain whether this finding is due to a decreased pulmonary vascular resistance and right ventricular pressure in the apelin-treated rats or is a direct effect on the right ventricular myocardium.
Therapeutic potential of apelin-APLNR in pulmonary hypertension
Apelin—a new modulator of pulmonary vascular resistance?
The acute vasodilatory effect of apelin on pulmonary arteries[47,108] and pulmonary pressure[109] is modest (10%-17%), and is attenuated in pulmonary hypertension. However, this does not exclude the possibility that vasodilatation might occur over a longer period with chronic administration. Apelin could potentially modify secretion of endothelial derived vasoactive factors, as shown in cardiomyocytes.[23] At any rate, current vasodilatory drugs available in the treatment of PAH are not sufficient to control the disease,[10] and focus in the management of PAH is now directed towards the basic mechanisms leading to proliferation and pulmonary vascular remodeling. The pressure-reducing effect seen in the long-term experimental models of pulmonary hypertension with chronic administration of apelin[16,23,110] may possibly be attributed to the stabilizing effect of apelin on endothelial cells,[16] and the prevention of loss of microvasculature.[48] Apelin may also affect inflammatory processes, although results are so far sparse and somewhat controversial.[40,118–121]
A new positive inotropic agent?
The benefits of the positive inotropic effect of apelin are uncertain. In heart failure caused by a compromised left ventricle, there are still no proven beneficial effects of any positive inotropic agents on survival despite many clinical studies with several drugs.[122] Actually, worse outcomes have been observed with β-adrenoceptor stimulating drugs and phosphodiesterase 3 inhibitors,[123] probably because of increments in energy expenditure and an increased propensity for cardiac arrhytmias, due to increased cAMP in the cardiac conductivity cells.[124] Even so, the search for useful positive inotropic agents is still ongoing, and the key to a successful drug may lie within the mechanism of which contractility is induced.[122] The known inotropic mechanisms for apelin differs from those of β-adrenoceptor agonists and phosphodiesterase 3 inhibitors, in that apelin has not been shown to increase levels of cAMP and levels of intracellular calcium may be unaffected.
Speaking in favor of apelin as a promising positive inotropic agent in chronic heart failure are the attenuating effects of angiotensin-II-induced cardiac fibrosis[100] and the fact that it prevents cardiac apoptosis in relation to ischemia.[33,115,117]
Potential adverse effects and pharmacological challenges
Apelin increases electrical conduction velocity in the heart[114] and apelin levels are changed in patients with cardiac arrhytmias.[125–127] Of 10 sheep injected with apelin, 4 developed some degree of atrio-ventricular block or other abnormalities of the electrocardiogram.[102] Therefore, the effects of apelin on cardiac conductivity and arrhytmias need closer human testing before clinical application.
Furthermore, obvious challenges for apelin as a drug for treatment of pulmonary hypertension are the short half life and the systemic vasodilatory effect, because PAH patients are already susceptible to symptomatic hypotension.[128] This may be overcome by pharmacological methods, e.g., chemical modification of the peptide and targeted delivery to the lungs by inhalation. Synthetic forms of apelin with the same biological activity as apelin-13 already exist,[129] and further development of such analogues may make the modulation of APLNR easier in the future, and allow different routes of administration.
CONCLUSIONS
In summary, apelin and APLNR are present in the pulmonary vasculature, are regulated by factors involved the pathogenesis of PAH, and are down-regulated in pulmonary arterial endothelial cells in patients with PAH. It may not, however, be suited as a specific biomarker for PAH.
Apelin and APLNR play a role in regulation of endothelial and smooth muscle cell homeostasis, and have effects similar and opposite to VEGF. Apelin modulates eNOS expression, induces eNOS-dependent vasodilatation in the systemic and pulmonary circulation, counteracts angiotensin-II-induced vasoconstriction, and has positive inotropic and cardioprotective effects (Fig. 4).
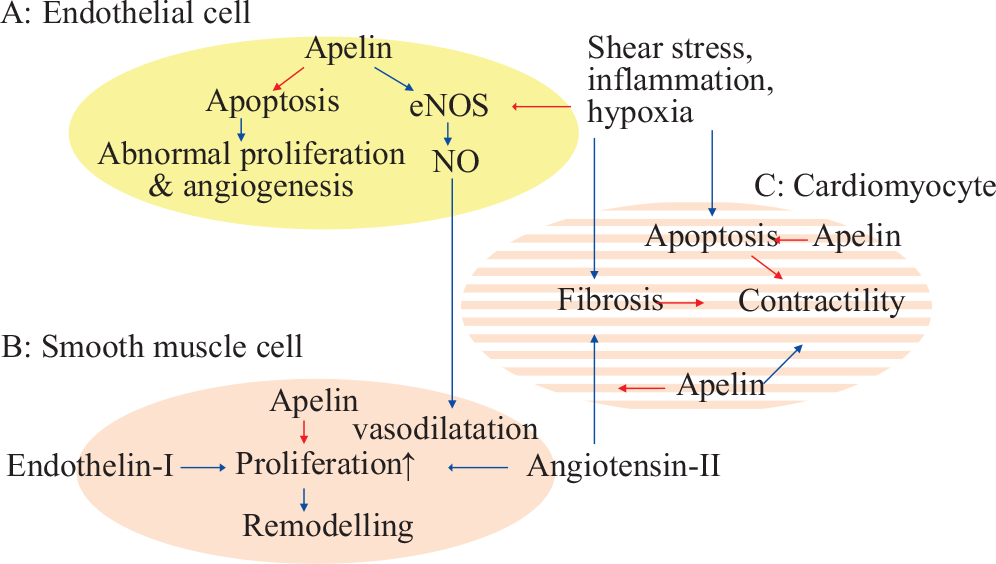
Factors in PAH pathogenesis opposed by apelin, A: In endothelial cells, B: In smooth muscle cells, and C: Right ventricle. Blue →: Stimulates activity/expression. Red →: Decreases activity/expression.
Thus in the laboratory, the peptide has many intriguing characteristics, but so far most studies have investigated effects in the left heart and systemic circulation, and the peptide has been administered to human subjects in only a very few studies. It should, however, be emphasized that the existing studies in experimental models of pulmonary hypertension point toward a beneficial effect of the drug (Table 2). Further research is needed to clarify the pharmacodynamics, pharmacokinetics and safety of apelin in pulmonary hypertension.
Studies on apelin in isolated pulmonary arteries, PAH patients or PAH animal models
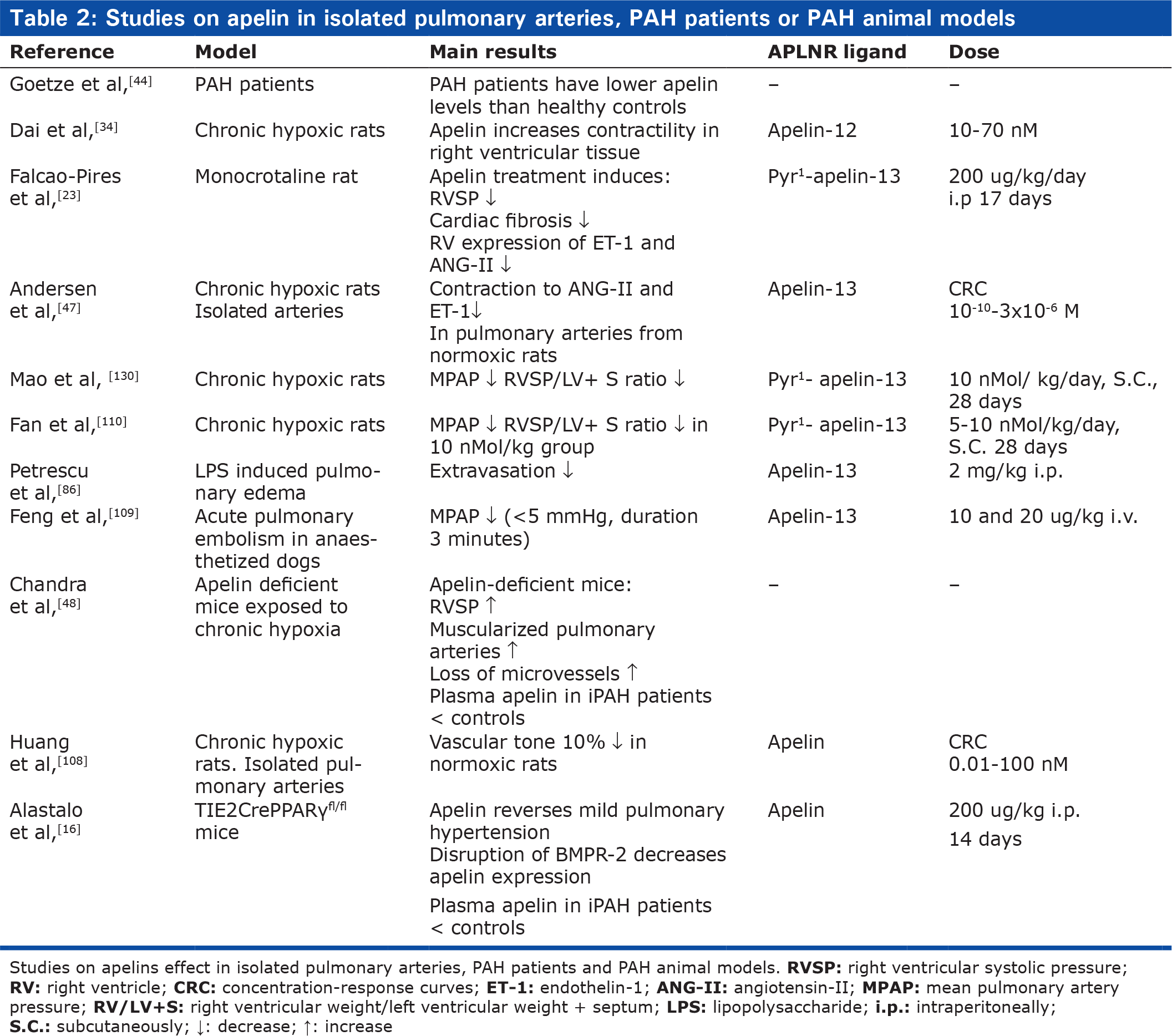
Studies on apelins effect in isolated pulmonary arteries, PAH patients and PAH animal models.