
Abstract
Pulmonary hypertension (PH) is a serious and progressive disorder that results in right ventricular dysfunction that lead to subsequent right heart failure and death. When untreated the median survival for these patients is 2.8 years. Over the past decade advances in disease specific medical therapy considerably changed the natural history. This is reflected in a threefold decrease in the number of patients undergoing lung transplantation for PH which used to be main stay of treatment. Despite the successful development of medical therapy lung transplant still remains the gold standard for patients who fail medical therapy. Referral for lung transplant is recommended when patients have a less than 2-3 years of predicted survival or in NYHA class III or IV. Both single and bilateral lung transplants have been successfully performed for PH but outcome analyses and survival comparisons generally favor a bilateral lung transplant.
INTRODUCTION
Pulmonary hypertension (PH), an abnormal elevation in pulmonary artery pressure, is defined as a mean pulmonary artery pressure ≥ 25 mmHg at rest or ≥ 30 mmHg with exercise, with a pulmonary capillary wedge pressure ≤ 15 mmHg as measured by cardiac catheterization.[1] PH was traditionally divided into primary and secondary but this classification system has since been replaced by a system proposed by the World Health Organization in 1998 and most recently updated at Dana Point, California in 2008.[2] The current classification system categorizes PH into five major categories with further subdivisions in each category allowing patients to be placed in groups sharing similarities in clinical presentation, pathophysiology, and therapeutic approaches (Table 1). Regardless of its etiology, PH is a serious and progressive disorder that results in right ventricular dysfunction and impairment in activity tolerance that can lead to subsequent right-heart failure and death.
Revised World Health Organization classification of pulmonary hypertension, Dana Point, California

Adapted from Simonneau et al.
Pathobiology
PAH has a multifactorial pathophysiology.[3] Abnormalities in molecular pathways regulating the pulmonary vascular endothelial and smooth muscle cells have been described as underlying PAH with perturbations in vasoconstriction, smooth-muscle cell and endothelial-cell proliferation, and thrombosis. This includes inhibition of the voltage-regulated potassium channel,[4] mutations in the bone morphogenetic protein-2 receptor,[5] increased serotonin uptake in the smooth muscle cell,[6] increased angiopoietin expression in the smooth muscle cells,[7] and excessive thrombin deposition related to a procoagulant state.[8] As a result, there appears to be a loss of apoptosis of the smooth muscle cells allowing their proliferation, and the emergence of apoptosis-resistant endothelial cells that can obliterate the vascular lumen. Vasoconstriction, remodeling of the pulmonary vessel wall, and thrombosis contribute to increased pulmonary vascular resistance in PAH. Pulmonary vascular remodeling occurs at all levels of the vessel wall. Endothelial cells, smooth muscle cells, and fibroblasts as well as inflammatory cells and platelets may play a significant role in PAH.
Pulmonary vasoconstriction is believed to be an early component of the pulmonary hypertensive process and may be related to abnormal function of potassium channels and endothelial dysfunction.[4] Endothelial dysfunction leads to chronically impaired production of vasodilators such as nitric oxide and prostacyclin along with overexpression of vasoconstrictors such as endothelin. [9] Recent genetic and pathophysiologic studies have emphasized the relevance of several mediators in this condition, including prostacyclin,[10] nitric oxide,[11] endothelin,[12] angiopoietin,[7] serotonin,[13] and members of the transforming growth factor superfamily (TGF)-β.[5]
Pathophysiology
The pathophysiology of PH can be understood as a lethal cycle of increased pulmonary vascular resistance (as a result of any of the causes listed in the WHO classification scheme) leading to increased right ventricular performance and oxygen consumption with resultant right ventricular hypertrophy and dilatation, leading to decreased cardiac output and eventual right ventricular failure (Fig. 1).[14,15] In response to an increase in resistance within the pulmonary circulation, the right ventricle responds by increasing right ventricular systolic pressure as necessary to preserve cardiac output. Over time, the pulmonary vascular system responds with progressive remodeling that sustains and promotes PH.
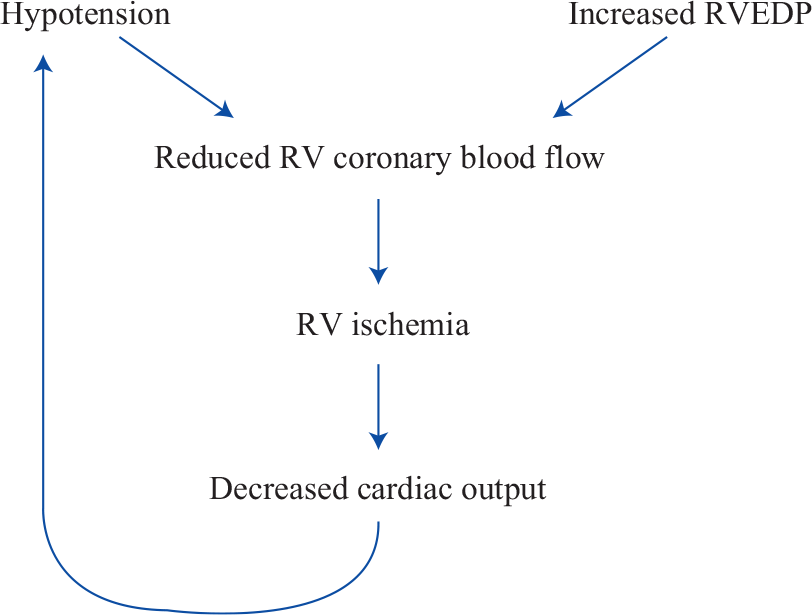
The downward spiral of pulmonary hypertension (adapted from Vigneswaran et al.).
The degree to which the right ventricle responds to such changes is dependent upon the age of the patient and rapidity of onset of PH. A large acute pulmonary embolism can result in right ventricular failure and shock whereas chronic thromboembolic disease of equal severity may result in only mild exercise intolerance.
The right ventricle is well designed to adapt to wide variations in preload owing to its anatomical structure and geometry, however these features are not suited to adequately deal with increases in afterload. One of the key features to right ventricular adaptation to chronic pressure overload is hypertrophy due to increased wall stress (Laplace's Law).[15] Hypertrophy is greatest in the right ventricular outflow tract and worse in patients with decompensated function.[15] In the setting of increased afterload, right ventricular stroke volume decreases linearly with increasing resistance leading to eventual ventricular dilatation and consequent decreased right ventricular coronary blood flow at a time when oxygen consumption is increased.[16] Furthermore, right ventricular dilatation shifts the interventricular septum to the left, decreasing left ventricular preload and compliance and thus cardiac output. Recent data also suggests that hypoxemia may impair the ability of the right ventricle to make compensatory changes. These studies suggest that right ventricular failure occurs in PH when the myocardium becomes progressively ischemic due to excessive demands and inadequate right ventricular coronary blood flow.[16] The onset of peripheral edema and other clinical manifestations of right heart failure usually portend a poor outcome.[17]
Presenting signs and symptoms
Patients with PH may present with a myriad of cardiopulmonary symptoms however exertional dyspnea is the most frequent symptom and unexplained dyspnea should always raise suspicion. PH may be asymptomatic in the early stages and may be an incidental finding on echocardiogram. Chest pain and syncope are usually late symptoms. Patients may present signs and symptoms of right heart failure such as peripheral edema or ascites. A family history of PH, use of fenfluramine appetites suppressants, cocaine or amphetamines, prior history of deep vein thrombosis (DVT) or pulmonary embolism (PE), chronic liver disease or portal hypertension, HIV, thyroid disease, splenectomy, and sickle cell disease should be sought in all patients suspected to have PH. Physical exam findings include increased jugular venous pressure, a reduced carotid pulse, and a palpable right ventricular impulse. Most patients have an increased pulmonic component of the second heart sound, a right-sided fourth heart sound, and tricuspid regurgitation. Peripheral cyanosis and/or edema tend to occur in later stages of the disease.
Diagnosis and assessment of functional status
The goals of work-up in PH include confirmation of diagnosis, establishing an underlying cause, and quantifying severity with hemodynamics and functional impairment. All patients who appear to have PH after non-invasive testing should undergo right heart catheterization confirm the diagnosis, quantify the degree of hypertension (measurement of pulmonary artery pressure, cardiac output, and left ventricular filling pressure, underlying cardiac shunt) and undergo acute vasodilator testing.
Acute vasodilator testing, during catheterization defines the extent of pulmonary vasodilator reactivity and dictates prognosis and therapy. The majority of centers use inhaled nitric oxide (NO) as a pulmonary vasodilator (10-80 ppm).[18] A positive vasodilator response is defined as a decrease of at least 10 mmHg in mean PAP and achieving a mean PAP <40 mmHg, an increase or no change in cardiac output with no significant fall in blood pressure.[19] Patients who respond to acute vasodilator therapy can often be treated with calcium channel blockers and have a more favorable prognosis.[20] Echocardiography is helpful in confirming the diagnosis and excluding left-sided lesions as the cause of PH but is not specific enough to confirm a diagnosis of PH alone. Overall functional status assessed by symptoms (New York Heart Association [NYHA] functional class/World Health Organization functional class [WHO-FC]) and functional capacity assessed primarily by the six-minute walk test (6-MWT) – a very simple and reproducible submaximal exercise test that can be performed by a patient not tolerating maximal exercise tests. The 6-MWT has been the cornerstone functional test to evaluate treatment efficacy both in clinical trials and in daily clinical practice. In unselected patients (treated or not), a 6-MWT of less than 332 meters is associated with a worse prognosis in iPAH and has a strong independent association with mortality in patients with type 1 PH.[21,22]
Prognosis
The natural history of PH is dismal, with a reported median survival rate of 2.8 years when untreated.[23] Functional class remains a strong predictor of survival, with patients who are in NYHA functional class IV having a mean survival of less than six months. The cause of death is usually right ventricular failure, which is manifest by progressive hypoxemia, tachycardia, hypotension, and edema. Over the past decade, advances in medical therapy considerably changed the prognosis of the disease. Most expert centers discuss the notion of transplantation early after diagnosis and closely follow patients' symptoms, functional status – including the six minute walk test distance, and hemodynamics.[24] Benza et al. analyzed data from 2716 patients with PAH in the US Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) study to determine factors that may predict 1-year survival in patients with PAH awaiting transplantion. The authors found in a multivariable analysis that factors independently associated with increased mortality included pulmonary vascular resistance >32 Wood units, PAH associated with portal hypertension, a modified NYHA/WHO functional class IV, men >60 years of age, and family history of PAH.[25] Their data also confirmed an increased mortality risk in patients with renal insufficiency or any pericardial effusion on echocardiogram.[25]
Medical management of PH
Of all the conditions for which lung transplantation is performed, PH is the only one in which there have been significant strides made in medical management. This is seen by the ever-decreasing number of patients with PH who ultimately undergo transplantation. In 1990, approximately 10.5% of all lung transplants were for patients with PH whereas in 2001 only 3.6% of all lung transplants were performed in patients with this condition and most recently, 3.3% as reported by the ISHLT Transplant Registry in 2010.[26–27] Treatment of PH is individualized, based upon severity of functional impairment. As summarized in Table 2, there are currently eight FDA-approved therapies for WHO group 1 PAH.[28]
US Food and Drug Administration-Approved medications for pulmonary hypertension
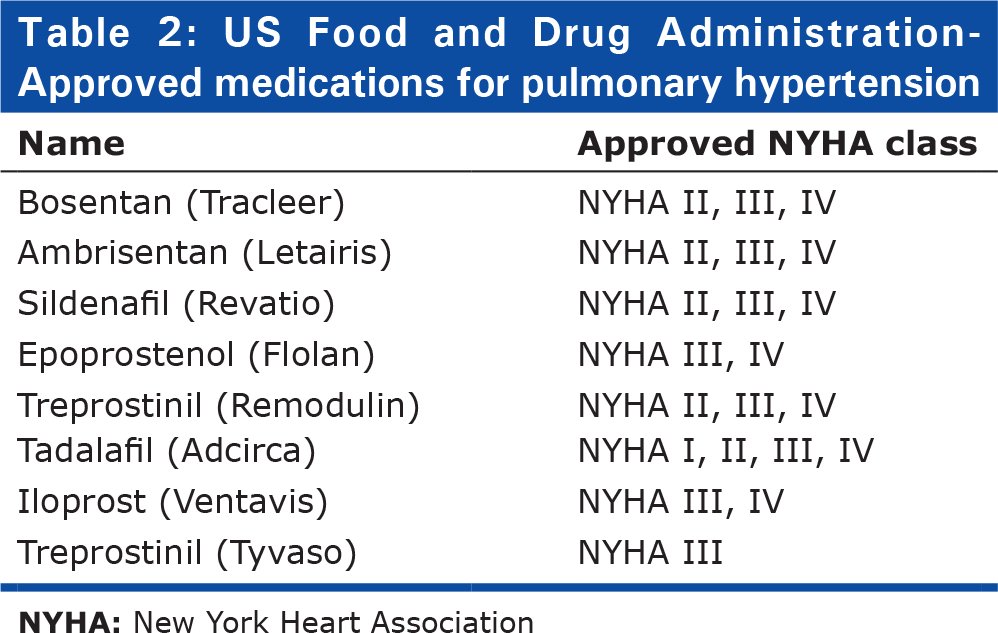
These medications include endothelin receptor antagonists (ERAs) (bosentatn and ambrisentan), phosphodiesterase-5 inhibitors (PDE5-I) (sildenafil and tadalafil), and prostanoid derivatives (epoprostenol, trepostinil, and iloprost). The latter group—the prostanoids introduced in the 1990s—have been the most important advance in the management of patients with PH. Chronic intravenous epoprostenol therapy leads to an improvement in exercise tolerance, hemodynamic measures, as well as survival in iPAH.[29,30] Initially intended to serve as a bridge to transplantation, with experience it has been realized that the need for transplantation can be averted in some cases. [31–34] Additionally, a select number of patients who have substantial reductions in pulmonary arterial pressure in response to short-acting vasodilators at the time of cardiac catheterization should be treated initially with calcium channel blockers.[20] Patients who respond favorably usually have dramatic reductions in pulmonary artery pressure and pulmonary vascular resistance associated with improved symptoms, regression of right ventricular hypertrophy and improved survival.[35] Anticoagulation with warfarin has been shown to provide a survival benefit in PH, even without documented thromboembolism. [36] Additionally, diuretic therapy relieves peripheral edema and may be useful in reducing right ventricular end diastolic pressure (RVEDP). Supplemental oxygen should be provided to alleviate dyspnea and right ventricular ischemia as hypoxemia is a potent pulmonary vasoconstrictor. A more comprehensive list of medical therapies is available in the most recent ACCP guidelines.[37]
Indications for lung transplantation
Despite the successful development of disease-specific medical therapies for PH which has reduced patient referral for lung transplant programs,[38] transplantation remains the gold-standard for patients who fail medical therapy. Survival among patients requiring treatment with intravenous prostacyclin is approximately 63% at 3 years[39,40] and up to 25% of patients with iPAH may fail to improve on disease-specific therapy and the prognosis of patients who remain in WHO-FC III or IV is poor.[32,33] McLaughlin et al. describe a treatment algorithm based on a risk assessment based on various clinical variables (Table 3). Patients at highest risk should be considered for intravenous therapy as first-line therapy and immediate assessment for lung transplantation. Patients as lower risk are candidates for oral therapy and should be followed closely, and response to therapy reassessed in several months if treatment goals are not met.[41]
Determinants of risk in patients with pulmonary hypertension
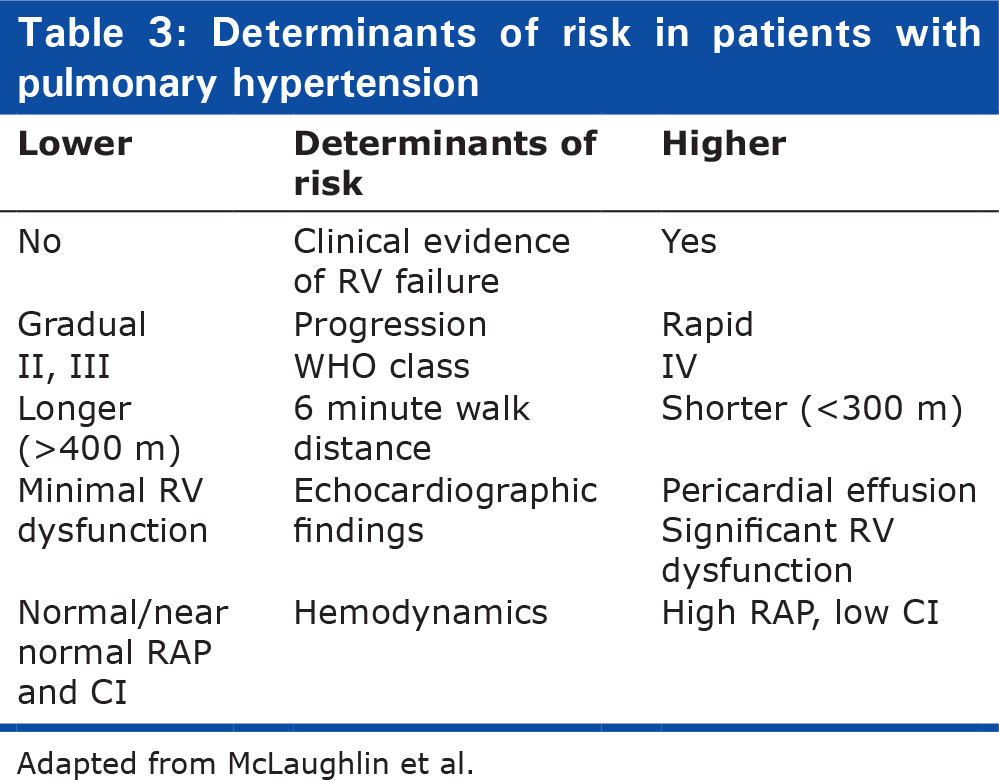
Adapted from McLaughlin et al.
In general, referral for transplantation assessment is advisable when patients have a less than 50%, 2- to 3- year predicted survival or NYHA class III or IV level of function, or both.[24] With regards to PAH, most experts recommend transplantation early after diagnosis based on patients' symptoms, functional status—including the 6-MWT distance—hemodynamics.[24] The decision to list for transplant is made when functional status and hemodynamics decline to the point where survival without transplantation is likely to be compromised[24] (Table 4).
ISHLT guidelines for lung transplantation

Type of transplantation
The appropriate surgical procedure for patients with iPAH and secondary PAH has been a topic of longstanding debate. Single lung transplant (SLT), bilateral lung transplant (BLT), and heart-lung transplant (HLT) are all used in the treatment of PH. HLT was originally the standard procedure for patients with PH until the 1990's and is still the preferred treatment for PH patients with irreversible heart disease. However the number of adult HLT have declined significantly in recent years[42–50] to about 70 to 90 per year. It has largely been replaced by bilateral lung transplant, now the most commonly used form of transplantation PH patients. This change was multifactorial: results with SLT or BLT for PH are comparable to or better than HLT, right heart dysfunction improves following SLT or BLT due to right ventricular recovery with the rapid reduction in pulmonary vascular resistance after lung transplantation, as well as ethical and pragmatic concerns centered on donor shortages and organ distribution.[51]
In 2008, according to the Official Lung and Heart-Lung Transplant Report of the Registry of the International Society for Heart and Lung Transplantation reported 73 heart-lung transplants and 2,769 lung transplant procedures. Among these, approximately one-quarter of heart-lung transplants were performed with PH as the primary diagnosis and while less than 5% of isolated lung transplant procedures were performed for PH.[52] Among patients undergoing isolated lung transplantation, BLT is used much more frequently-accounting for 91% of transplants performed for PH and the remaining 9% consisting of SLT.[52]
Outcomes following isolated lung transplantation for pulmonary hypertension
For all lung transplant recipients, overall unadjusted survival rates were 79% at one year, 63% at three years, 52% at five years, and 29% at 10 years.[52] Survival rates at three months after lung transplantation were lowest for iPAH (76%) as well as idiopathic pulmonary fibrosis (85%) and highest for cystic fibrosis (90%) and chronic obstructive pulmonary disease (91%).[52] In contrast, among patients surviving at least one year, diagnoses of iPAH, cystic fibrosis, sarcoidosis, and emphysema had significantly better survival at five and 10 years following lung transplantation than those with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis.
The evidence regarding the optimal transplant procedure for PH is remains weak. Available data is largely composed of single center studies. Both SLT and BLT have been performed successfully for PH however outcome analyses and survival comparisons generally favor BLT. There is a trend towards a survival benefit with BLT in the ISHLT registry, however it does not reach statistical significance.
Those who support SLT argue that SLT is easier to perform technically, has less morbidity and mortality versus BLT and HLT, less ischemic time and bypass time with resultant better early graft function, has improved early survival, and will allow more patients to receive lung transplants. Hemodynamics following SLT for PH are characterized by a rapid and sustained drop in pulmonary artery pressures, substantial improvement in right ventricular function, and a preponderance (>90%) of pulmonary blood flow to the allograft as a result of high pulmonary vascular resistance in the remaining native lung.[53–55] This leads to a postoperative situation in which ventilation is evenly divided between the allograft and native lung while perfusion is almost entirely directed to the allograft. Any complication in the allograft (e.g., pneumonia, primary graft dysfunction, rejection) can result in severe ventilation-perfusion mismatch and hypoxemia.
Most centers favor BLT because of the physiologically increased functional reserve, making patients less prone to respiratory insufficiency with subsequent insulti[47,56] Proponents of BLT have argued that bilateral lung transplants result in less ventilation perfusion (V/Q) mismatches, are easier to care for in the perioperative period, will enable more “marginal lungs” to be utilized, will provide better overall lung function, are protective against the physiologic manifestations of bronchiolitis obliterans, and have a better long-term survival. In its most recent publication, the ISHLT registry reported a significantly better survival in the bilateral lung transplantation group as compared with the single lung transplant in patients with iPAH.[52] The Pittsburgh group compared recipients of SLT versus BLT with PH and found similar functional status and postoperative recovery (hemodynamics, duration of mechanical ventilation, intensive care unit/hospital length of stay, and incidence of acute/chronic rejection).[43] They did, however, report that SLT recipients had a significantly lower PaO2/FiO2 at one hour and higher mean pulmonary artery pressure at 12 and 24 hours.[43] Bando et al. concluded that despite significantly shorter cardiopulmonary bypass duration in their cohort, SLT recipients with underlying PH had significantly less functional recovery (less reduction in mean pulmonary artery pressure and increase in cardiac index) and higher graft mortality compared with BLT and HLT recipients.[45] The Hopkins experience evaluated 15 patients with iPAH and found BLT to have a highly significant survival advantage compared with SLT at 4 years of follow-up.[51] The most recent ISHLT registry reported 788 iPAH lung transplant recipients of which 91% were BLT, highlighting the continuing trend at most centers favoring BLT versus SLT for iPAH.[27]
Outcomes following heart-lung transplantation for pulmonary hypertension
According to the 2009 ISHLT registry report comparing the outcome of HLT with BLT for pulmonary hypertension demonstrated similar survival between the two groups. Of 2712 adult patients who underwent heart-lung transplantation between 1982 and 2007 survival rates of 72 percent at three months and 64 percent at one year. Survival for HLT was less than with lung transplantation alone (89% and 79%, respectively, in the era from 1994 to 2007). However, overall survival five years and ten years was 43% and 28%, which was comparable to isolated lung transplantation. Among HLT patients who survived one year, there was a low but steady mortality rate with a survival conditional half-life (contingent on survival to one year) of 9.2 years.[57]
CONCLUSIONS
Lung transplant is an effective form of treatment for patients with PH who have exhausted all medical therapy BLT is the current choice of transplantation in this patient population.