
Abstract
Pulmonary vasoconstriction and vascular remodeling are two major causes for elevated pulmonary vascular resistance and pulmonary arterial pressure in patients with idiopathic pulmonary arterial hypertension (IPAH). An increase in cytosolic free Ca2+ concentration ([Ca2+]cyt) in pulmonary artery smooth muscle cells (PASMC) is a major trigger for pulmonary vasoconstriction and an important stimulus for PASMC proliferation, which causes pulmonary vascular remodeling. Store-operated Ca2+ entry (SOCE), induced by depletion of stored Ca2+ in the sarcoplasmic reticulum (SR), can increase [Ca2+]cyt in PASMC, independent of other means of Ca2+ entry. Stromal interaction molecule (STIM) proteins, STIM1 and STIM2, were both recently identified as sensors for store depletion and also signaling molecules to open store-operated Ca2+ channels. We previously reported that SOCE was significantly enhanced in PASMC from IPAH patients compared to PASMC from normotensive control subjects. Enhanced SOCE plays an important role in the pathophysiological changes in PASMC associated with pulmonary arterial hypertension. In this study, we examine whether the expression levels of STIM1 and STIM2 are altered in IPAH-PASMC compared to control PASMC, and whether these putative changes in the STIM1 and STIM2 expression levels are responsible for enhanced SOCE and proliferation in IPAH-PASMC. Compared to control PASMC, the protein expression level of STIM2 was significantly increased in IPAH-PASMC, whereas STIM1 protein expression was not significantly changed. In IPAH-PASMC, the small interfering RNA (siRNA)-mediated knockdown of STIM2 decreased SOCE and proliferation, while knockdown of STIM2 in control PASMC had no effect on either SOCE or proliferation. Overexpression of STIM2 in the control PASMC failed to enhance SOCE or proliferation. These data indicate that enhanced protein expression of STIM2 is necessary, but not sufficient, for enhanced SOCE and proliferation of IPAH-PASMC.
INTRODUCTION
Increased pulmonary vascular resistance, due to sustained vasoconstriction, vascular remodeling, in situ thrombosis, and increased vascular wall stiffness, is the major cause for elevated pulmonary arterial pressure in patients with pulmonary arterial hypertension (PAH).[1,2] Studies from a pulmonary angiogram show that patients with idiopathic pulmonary arterial hypertension (IPAH) and hypoxia-induced pulmonary hypertension (HPH) have a significant decrease in the blood flow to small- and medium-sized pulmonary arteries. Decreased blood flow to small and medium-sized pulmonary arteries results mainly from a decrease in the diameter of the artery lumen, due to sustained pulmonary vasoconstriction and vascular remodeling — two major causes that lead to increased pulmonary vascular resistance and pulmonary hypertension. HPH and IPAH share many pathological and histological traits, such as, concentric vascular remodeling and medial hypertrophy. In fact, rats subjected to chronic hypoxia are used as in vivo models for studying the pathogenic and therapeutic mechanisms of pulmonary arterial hypertension, and rat pulmonary artery smooth muscle cells (PASMC) treated with hypoxia are common in vitro models for studying the cellular and molecular sequences of events involved in pulmonary vascular remodeling. Pulmonary vascular remodeling due to excessive proliferation of PASMC and sustained pulmonary vasoconstriction due to contraction of PASMC contribute greatly to the elevated pulmonary vascular resistance in patients and animals with IPAH and HPH.
An increase in cytosolic Ca2+ concentration ([Ca2+]cyt) in PASMC is a major trigger for pulmonary vasoconstriction and an important stimulus for cell proliferation and migration that contributes to pulmonary vascular remodeling. Removal of extracellular Ca2+ or reduction of extracellular free [Ca2+] with the Ca2+ chelator (e.g., EGTA and EDTA) not only significantly inhibits agonist-induced vasoconstriction in the isolated pulmonary arterial rings, but also significantly attenuates PASMC proliferation when cultured in growth factor-containing media. Both blockade of Ca2+ influx and depletion of intracellular Ca2+ stores in the sarcoplasmic reticulum (SR) or endoplasmic reticulum attenuate PASMC contraction and proliferation. Our preliminary studies show that resting [Ca2+]cyt is increased and that agonist-induced rise in [Ca2+]cyt is significantly enhanced in patients with IPAH when compared with normotensive control subjects.[3] Furthermore, chronic hypoxia also increases [Ca2+]cyt in PASMC.[4,5] Therefore, increased proliferation and contraction of PASMC in IPAH and HPH patients are likely related to the increase in [Ca2+]cyt and enhancement of the mechanisms that mediate Ca2+ influx.
Store-operated Ca2+ entry (SOCE) is an important mechanism that mediates Ca2+ influx and raises [Ca2+]cyt when the intracellular stores are depleted by agonist- or ligand-induced Ca2+ mobilization.[6,7] Depletion of intracellular Ca2+ stores, predominantly the sarcoplasmic reticulum (SR) in PASMC, activates a Ca2+ influx through the store-operated Ca2+ channels (SOC) expressed on the plasma membrane. Stromal interacting molecule (STIM) proteins, which include two isoforms, STIM1 and STIM2, are single transmembrane proteins that have been identified as the sensors of store depletion.[8,9] STIM1 and STIM2 are expressed on the SR membrane of PASMC. An EF-hand domain near the N-terminus of STIM1 and STIM2 serves as the sensor of Ca2+ concentration in the SR lumen. STIM1 and STIM2 are locked in an inactive conformation when Ca2+ is bound to the EF-hand domain, but when Ca2+ is depleted in the SR the conformation of STIM1 and STIM2 changes to active conformation.[9,10] The active STIM1 (and STIM2) can oligomerize and translocate to ER-PM junctions, activate ER-PM junctions Ca2+ channels, and elicit SOCE. In HEK cells, overexpression of STIM1 can reconstitute a robust inwardly rectifying Ca2+ current following store depletion.[11] Similar studies have shown that STIM2 can reconstitute SOCE in the absence of STIM1, although at a lower magnitude of Ca2+ influx.[12,13] While STIM2 has lower activity in activating Ca2+ channels (or store-operated Ca2+ channels) on the plasma membrane, it is more sensitive to small changes in SR Ca2+ concentration.[14,15]
Our preliminary data indicate that both receptor-operated and store-operated Ca2+ entry play an important role in normal PASMC proliferation and migration.[16,17] Inhibition of SOCE with, for example, Ni2+, 1-[β-[3-(4-methoxyphenyl)pro-poxy]-4-methoxyphenethyl]-1H-imidazole hydrochloride (SKF96365), or La3+, markedly attenuates PASMC proliferation.[3,18–20] Furthermore, SOCE, induced by the passive depletion of Ca2+ stores using cyclopiazonic acid (CPA), an inhibitor of Ca2+-Mg2+ ATPase (SERCA) on the SR membrane is significantly enhanced, and the mRNA and protein expression of the canonical transient receptor potential (TRPC) channels are upregulated in the PASMC from IPAH patients in comparison to normotensive controls.[21,22] We have previously shown that upregulated TRPC channels (e.g., TRPC3 and TRPC6) contribute in enhancing SOCE in IPAH-PASMC, it is not known whether STIM1 and / or STIM2 are also involved in enhancing SOCE in IPAH-PASMC. Specifically, it is not known whether the protein expression of STIM1 and / or STIM2 changes in IPAH -PASMC and whether any observed changes in protein expression are responsible for enhanced SOCE and proliferation in IPAH-PASMC. In this study, we compare the protein expression levels of STIM1 and STIM2 in IPAH-PASMC and control PASMC, and investigated the functional role of any putative changes in protein expression level of STIM 1 and STIM2 on SOCE and proliferation in PASMC.
MATERIALS AND METHODS
PASMC isolation and culture
Sprague-Dawley rats (125-250 g) were decapitated; the right and left branches of the main pulmonary artery as well as the intrapulmonary arteries and surrounding tissues were removed and placed in Hanks' solution (HBSS), comprised of Hanks' balanced salt (Irvine Scientific, Santa Ana, CA) supplemented with 14.98 mM N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid (HEPES), 4.17 mM NaH2CO3, 0.4 mM MgSO4, 10,000 U/ml penicillin, 10 mg/ml streptomycin, and 0.02 mM CaCl2. The connective tissues were gently removed under a dissecting microscope using sterile conditions. The isolated PA was incubated in 1 ml HBSS containing 1.7 mg collagenase (Worthington Biochemical, Freehold, NJ) for 20 minutes. The adventitia was carefully stripped off and the endothelium was scraped off with a fine forceps. The resulting PA smooth muscle tissue was incubated at 37°C for 20–25 hours in 10% fetal bovine serum culture medium (FBSCM), comprised of Dulbecco's modified Eagle's medium (DMEM, Sigma Chemical, St. Louis, MO) supplemented with 7 mM NaH2CO3 and 10 mM HEPES at pH 7.2, and fortified with fetal bovine serum (10%, Irvine Scientific). The overnight incubation of the smooth muscle tissues in 10% FBSCM, before enzymatic digestion, improved the yield of the cells. The tissue was then incubated with 1.7 mg collagenase (Worthington), 0. 5 mg elastase (Sigma), and 1 mg bovine serum albumin (Sigma) in 1 ml HBSS at 37°C for 40 minutes. After 15–20 minutes, the tissue was triturated three to five times with a fire polished Pasteur pipette, to speed the digestion. The incubation mixtures were then diluted 20-fold by adding 20% FBSCM to stop the digestion. The cell suspensions were centrifuged for 5 minutes at 1,500 rpm, at room temperature, and the supernatant was removed. The resulting pellets were resuspended in 2–3 ml of 10% FBSCM and triturated with fire-polished Pasteur pipettes to separate the cells. Aliquots (3-6 drops) of the cell suspensions were drawn off and placed on cover slips in Petri dishes with 2 ml of 10% FBSCM. The cells were fed every 48 hours with 10% FBSCM and incubated in a humidified atmosphere of 5% CO, in air, at 37°C for three to seven days. After 10–20 days, the primary cultured cells had reached a confluence on the cover slips. At this time, the cells were treated with trypsin (1 mg/ml, Sigma) in HBSS and then replated on the cover slips in Petri dishes, with the addition of 2 ml of 10% FBSCM. Six to twelve hours later, the 10% FBSCM in the Petri dishes was replaced by fresh 10% FBSCM, to remove the dead and unattached cells.
Human PASMC were isolated from the lobectomized or resected lungs of normotensive control patients and from explanted lungs of patients with IPAH. The mean pulmonary arterial pressure of the IPAH patients was 52 mmHg. Patients undergoing lobectomy for bronchogenic carcinoma, who had no evidence of pulmonary hypertension by physical examination, electrocardiogram (ECG), echocardiogram, or pathological examination of resected lung tissue, and four patients with obstructive disease, who had normal pulmonary arterial pressures, were the sources of tissue for the normotensive control experiments. Lung tissue, removed from patients in the operating room, was immediately placed in cold (4°C) saline and taken to the laboratory for dissection. The muscular pulmonary arteries were incubated in Hanks' balanced salt solution (20 minutes) containing 2 mg/ml collagenase (Worthington Biochemical). The adventitia was stripped, and the endothelium removed. The remaining smooth muscle was digested with 2.0 mg/ml collagenase, 0.5 mg/ml elastase, and 1 mg/ml bovine albumin (Sigma Chemical Co.) at 37°C, to make a cell suspension of PASMC. The cell suspension was plated onto 25 mm cover slips, and incubated in a humidified atmosphere of 5% CO2 in air, at 37°C, in 10% fetal bovine serum, DMEM, for one week. The cells were isolated from arteries that were between 100–500 μm in diameter.
After isolation, all PASMC were cultured in M199 supplemented with 10% FCS, 100 μg/ml endothelial cell growth supplement (ECGS), 100 IU/ml penicillin, 100 μg/ml streptomycin, and 50 mg/L D-valine. The cells were plated on the cover slips and coated with 2% gelatin. Approval to use the human lung tissues and cells was granted by the University of California, San Diego (UCSD) Institutional Review Board.
All cells were incubated in a humidified environment at 37°C and 5% CO2. The medium was changed 24 hours after initial seeding and every 48 hours subsequently. When the cells reached 80-90% confluency, they were gently washed with phosphate buffered saline (PBS), incubated briefly with 3 mL of 0.025% trypsin / EDTA solution until detachment (3-5 minutes), and then incubated with an equal amount of trypsin neutralizing solution or serum. The cell suspension was then transferred to a sterile 15 ml round bottom tube, centrifuged at room temperature for 5 minutes, at 200 times gravity, and then resuspended in the appropriate growth media and seeded. Human PASMC between passages 3 and 8 were used for experiments, and rat PASMC were used during the first passage.
Measurement of cytosolic Ca2+ concentration ([Ca2+]cyt)
Cells on 25 mm cover slips were placed in a recording chamber on the stage of an inverted Nikon Eclipse / TE 200 microscope with the TE-FM epifluorescence attachment. [Ca2+]cyt was measured in each cell using the membrane-permeable Ca2+-sensitive fluorescent indicator, Fura-2-AM (Invitrogen). The cells were incubated at room temperature for 30 minutes in modified Krebs solution (MKS) containing 4 μM Fura-2-AM. The loaded cells were then washed with modified Krebs solution (MKS) for 30 minutes, to remove the excess extracellular dye and to allow the intracellular esterases to cleave the cytosolic Fura-2-AM into the active Fura-2 that was trapped in the cytosolic compartment. Fura-2 fluorescence was observed as a 510-nm-wavelength light emission with excitation wavelengths of 340 and 380 nm, with a use of the digital fluorescence imaging system from Intracellular Imaging. In all the experiments, multiple cells were imaged in a single field, and one arbitrarily chosen peripheral cytosolic area from each cell was spatially averaged. [Ca2+]cyt was expressed as a Fura-2 fluorescence emission ratio, excited at 340 and 380 nm (F340/F380), and then normalized to initial F340/F380 (F/F0).
The MKS had an ionic composition of 140 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 10 mM glucose, and 10 mM HEPES. The pH was adjusted to 7.4 with 10 N NaOH. The recording chamber was continuously perfused with MKS at a flow rate of 2 ml / minute using a mini-pump (Model 3385; Control, Friendswood, TX). The [Ca2+]cyt measurements were carried out at 32°C using an automatic temperature controller (TC-344B, Warner Instruments, Holliston, MA).
Western blot
The cells were lysed (Lysate buffer: 0.5% sodium deoxycholate, 0.1% SDS, 1% triton-X, 0.1% protease inhibitor cocktail) and centrifuged at 16,000 g for 10 minutes at 4°C. The supernatants were used as protein samples. The samples were separated through an SDS-polyacrylamide gel and transferred to the nitrocellular membranes. The membranes were incubated with anti-STIM1, STIM2, antibodies (1:1,000), and followed by a secondary antibody (1:4,000) application. The immunoblots were detected with the ECL Western blotting detection reagents (Perkin-Elmer, Norton, OH). Band intensity quantified with ImageJ64, normalized to β-tubulin control, and expressed as arbitrary units. All antibodies were purchased from Pro-Sci or Sigma.
Transfection of PASMC
Pulmonary artery smooth muscle cells were transfected with siRNA against STIM2 (Santa Cruz sc-76589) or scramble siRNA-A (Santa Cruz sc-37007) as control using the Amaxa Nucleofector electroporation system. Sixteen microliters of a 10 μM solution of siRNA was used per 2 × 105 cells, in each reaction. Overexpression of STIM2 (Plasmid 18868: pEX-CMV-SP-STIM2 (15-746)) was performed by the Amaxa Nucleofector electroporation system. The STIM2 plasmid was ordered from Addgene, as provided by Dr. Tobais Meyer. Protein expression was examined, SOCE was measured, and cell proliferation was determined by cell counting, 48 hours following transfection.
PASMC proliferation assay
Pulmonary artery smooth muscle cells proliferation was determined by counting cells using the Bio-Rad TC10 Automated Cell Counter. Forty-eight hours after treatment (with siRNA or cDNA), the PASMC were counted and divided into three groups, with equal number of cells. These three groups of PASMC were then re-plated (0 hour) and counted after 24, 48, and 72 hours, respectively.
Statistical analysis
The composite data are expressed as mean±SE. Statistical analysis was performed using the unpaired Student's t-test. Differences were considered to be significant when *P<0.05, **P<0.01, and ***P<0.005.
RESULTS
Higher expression levels of STIM2, but not STIM1, in PASMC from IPAH patients
As previously described, PASMC from IPAH patients show enhanced SOCE compared to control PASMC.[18,22,24] However, it is not known whether the expression level of the proteins that mediate SOCE by sensing store depletion is altered in the PASMC from IPAH patients. PASMC were isolated and cultured either from non-pulmonary hypertensive patients (NPH-PASMC) or patients with IPAH (IPAH-PASMC). The protein levels of STIM1 and STIM2 were examined in IPAH-PASMC and NPH-PASMC whole cell lysate. In order to compare the protein expression levels of STIM1 and STIM2 between IPAH-PASMC and NPH-PASMC, we measured the protein concentration in the whole cell lysates and standardized protein loading with β-tubulin as a loading control. As shown in Figure 1a, there was no significant morphological difference between NPH-PASMC and IPAH-PASMC. The protein expression level of STIM2, however, was significantly upregulated in IPAH-PASMC compared to NPH-PASMC [Figure 1b and c]. Surprisingly, the STIM1 protein expression level was not significantly changed in IPAH-PASMC; however, it seemed that the level of the STIM1 protein expression had a trend to be lower in IPAH-PASMC than in NPH-PASMC [Figure 1c]. These data suggest that STIM2, rather than STIM1, may be important in enhancing SOCE in PASMC from IPAH patients, in addition to the upregulated TRPC channels, which we previously reported.[21,22]
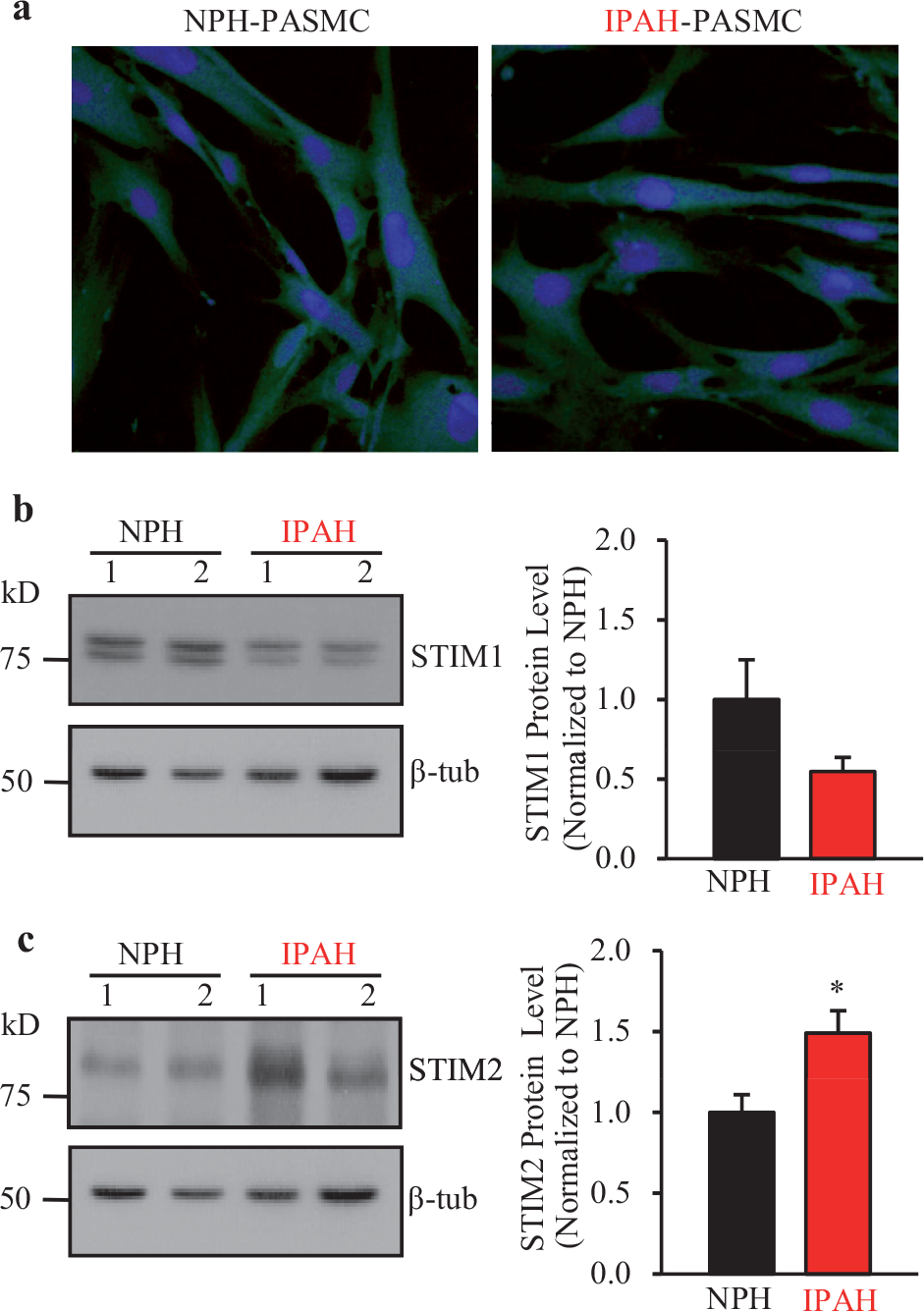
Protein expression of STIM2, but not STIM1, is increased in IPAH patients' PASMC. (a) Photograph showing that PASMC isolated from normotensive control subjects (NPH) and IPAH patients are morphologically comparable. (b and c) Representative Western blot images (left panels) and summarized data (right panels) for STIM1 (b) and STIM2 (c) proteins in PASMC isolated from IPAH patients' lung tissues or normotensive control patient lung tissues (NPH). β-tubulin was used as a loading control. Summarized data of STIM1 (b, right panel) or STIM2 (c, right panel) protein expression level (mean±SE) in IPAH-PASMC (n=6) and NPH-PASMC (n=6). Graph shows protein expression of STIM1 and STIM2 normalized to an average level in NPH-PASMC. *P<0.05 versus NPH. STIM2 protein expression level was increased in IPAH-PASMC, but STIM1 protein expression level was not changed in IPAH-PASMC
Decreasing protein expression level of STIM2 in PASMC from IPAH patients with siRNA
In order to further examine the functional role of STIM2 in PASMC from IPAH patients, small interfering RNA (siRNA) was used to decrease the protein expression level of STIM2 in PASMC. IPAH-PASMC were transfected with different concentrations (4, 8 or 16 μl) of 10 μM siRNA targeting STIM2 (siSTIM2) or 16 μl of 10 μM scrambled siRNA (control siRNA). Protein expression levels of STIM2 were examined with Western blot 48 hours following transfection. FACS analysis showed over 75% transfection efficiency with Enhanced Green Fluorecence Protein (EGFP) in PASMC (data not shown). siSTIM2 significantly decreased protein expression of STIM2 in IPAH-PASMC [Figure 2]; 4, 8, and 16-μl siSTIM2 decreased STIM2 protein expression levels in IPAH-PASMC by roughly 20, 60, and 80%, respectively, compared to scrambled siRNA control [Figure 2a and b]. The 16 μl dose of 10 μM of siSTIM2 and scrambled siRNA (control siRNA) was chosen as the dose to knockdown STIM2 in the experiments shown a little later in the text.
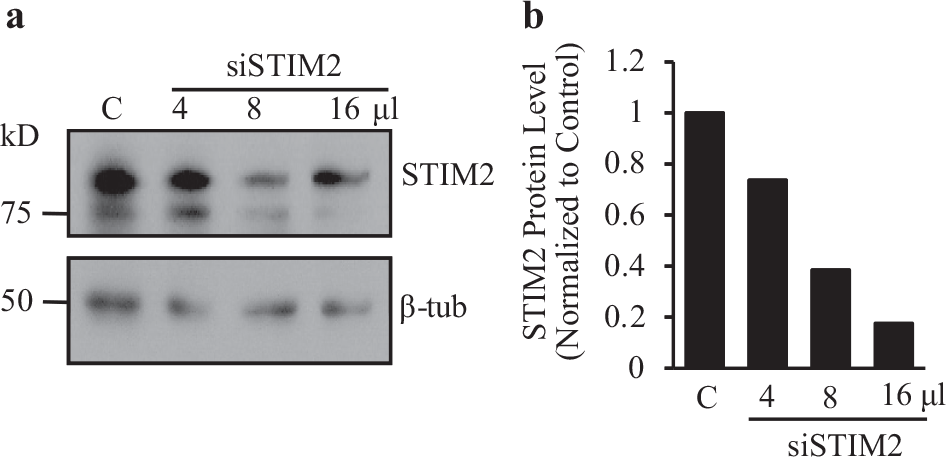
Dose-dependent knockdown of STIM2 in IPAH patients' PASMC with siRNA. (a) Representative Western blot image of STIM2 protein in IPAH-PASMC treated with siRNA against STIM2 (siSTIM2; with doses of 4, 8, and 16 μl) or control scrambled siRNA (indicated by ‘C’). β-tubulin (β-tub) was used as a loading control. (b) Quantification of STIM2 protein expression level in IPAH-PASMC treated with control scrambled siRNA (‘C’) or 4, 8, and 16 μl of siRNA-STIM2. Values were normalized to the β-tub level at the start and then normalized to the level of cells treated with control scrambled siRNA. The STIM2 protein expression level in IPAH-PASMC was decreased in a dose-dependent manner by siRNA targeting STIM2
STIM2 is necessary for enhanced SOCE in PASMC from IPAH patients
Change in [Ca2+]cyt was measured as the ratio of F340 / F380 (F) and normalized to the initial F340/ F380 ratio (F0), measured in PASMC superfused with physiological salt solution (PSS). SOCE was induced by the passive depletion of SR Ca2+ using 10 μM cyclopiazonic acid (CPA) in the absence of extracellular Ca2+, followed by re-addition of 1.8 mM extracellular Ca2+. Consistent with our previously reported findings,[22,24] IPAH-PASMC exhibited significantly larger (P=0.010) SOCE than NPH-PASMC [Figure 3a and b]. The siRNA-mediated knockdown of STIM2 in IPAH-PASMC led to a significant decrease (P=0.023) in the amplitude of SOCE [Figure 3b and c]. There was no significant difference in the magnitude of SR Ca2+ release, or a rise in [Ca2+]cyt due to Ca2+ leakage from the SR in NPH-PASMC, IPAH-PASMC treated with scramble siRNA, or IPAH-PASMC treated with siRNA-STIM2 (siSTIM2). Furthermore, there was no statistically significant difference (P=0.224) in SOCE between NPH-PASMC and IPAH-PASMC treated with siRNA-STIM2 [Figure 3b]. Western blot experiments also confirmed that the protein level of STIM2 was higher in IPAH-PASMC than in NPH-PASMC, and that siRNA-STIM2 effectively decreased the protein expression level of STIM2 in IPAH-PASMC [Figure 3c]. These data suggest that the increased protein expression level of STIM2 is necessary for enhanced SOCE in IPAH-PASMC.
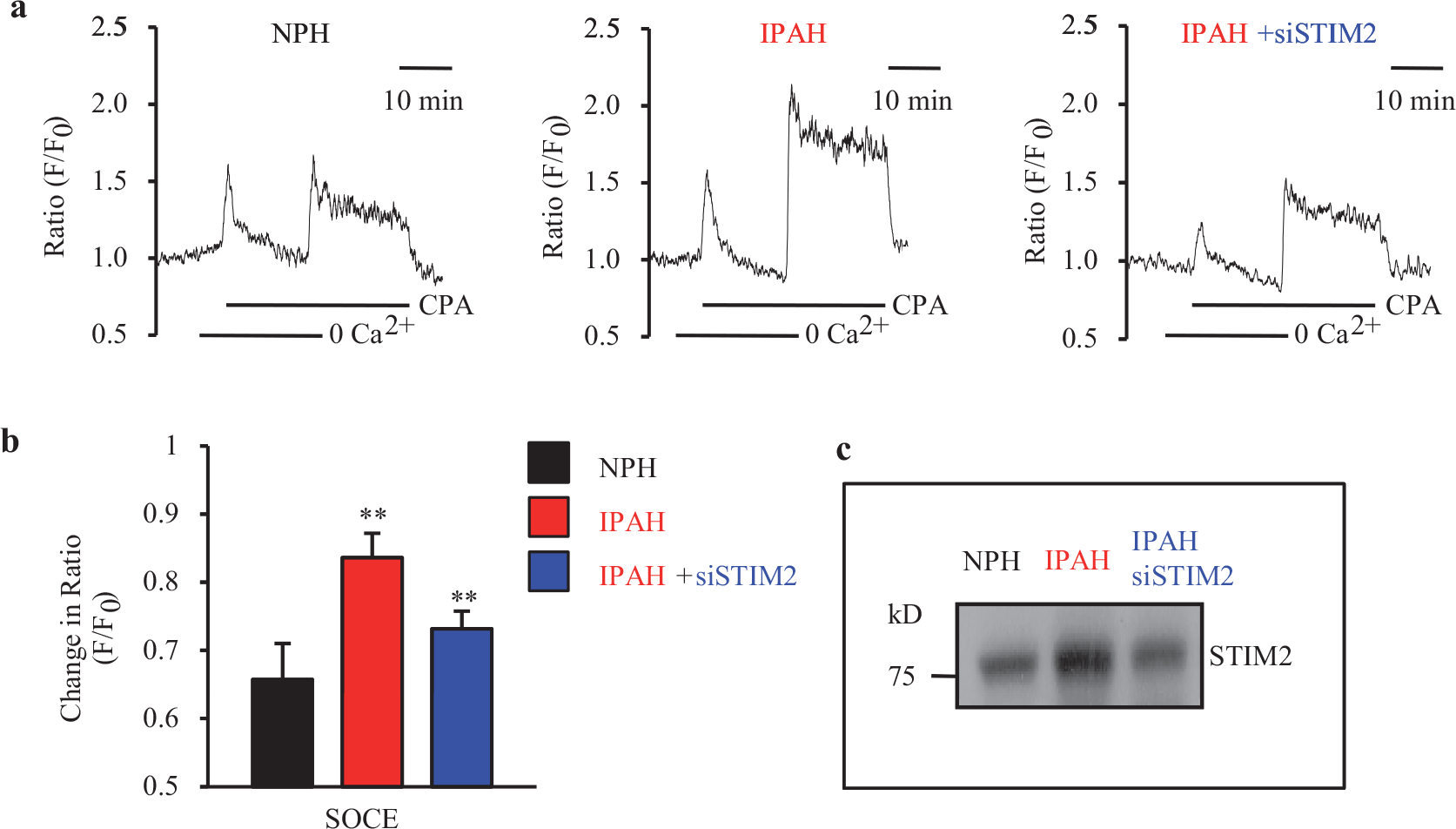
Upregulated protein expression of STIM2 is necessary for enhanced SOCE in IPAH patients' PASMC. (a) Representative records show CPA-induced changes in [Ca2+]cyt in the absence or presence of extracellular Ca2+ in NPH-PASMC (NPH, left panel), IPAH-PASMC (IPAH, middle panel), and IPAH-PASMC treated with siRNA targeting STIM2 (IPAH + siSTIM2, right panel). SOCE (indicated by the CPA-induced increase in [Ca2+]cyt when extracellular Ca2+ is restored) is induced by the passive depletion of SR Ca2+ using 10 μM CPA. NPH-PASMC (left panel) and IPAH-PASMC (middle panel) were treated with scrambled siRNA as a control. (b) Summary data (mean±SE) showing changes in CPA-induced increase in [Ca2+]cyt, immediately following the re-addition of Ca2+ after store depletion (indicative of SOCE) in NPH-PASMC (black bar), IPAH-PASMC (red bar), and IPAH-PASMC, treated with siRNA-STIM2 (blue bar). *P<0.01 (NS, not significant) versus NPH. (c) Western blot image of STIM2 protein expression in NPH-PASMC (NPH), IPAH-PASMC (IPAH), and IPAH-PASMC, treated with siRNA-STIM2 (IPAH + siSTIM2). NPH-PASMC (black) and IPAH-PASMC (red) were treated with scrambled siRNA
STIM2 is necessary for the increased proliferation of PASMC from IPAH patients
Proliferation of PASMC was determined by counting cells at the 0 hour (i.e., two days after the cells were plated onto the Petri dishes) and 24, 48, and 72 hours later. At the 0 hour, the control (NPH) PASMC group and IPAH-PASMC group had roughly the same number of cells [Figure 4a]. After 72 hours, the numbers of NPH-PASMC and IPAH-PASMC were both significantly increased [Figure 4a and b]; however, the increase in the IPAH-PASMC number was much greater that in the NPH-PASMC [Figure 4b]. Knockdown of STIM2 with siRNA had little effect on the change in the NPH-PASMC number after 72 hours [Figure 4c, left panel]. However, knockdown of STIM2 with siRNA in the IPAH-PASMC significantly decreased the increase in cell number after 72 hours (P<0.001) [Figure 4c, right panel, and d]. The siRNA-STIM2-mediated knockdown of STIM2 appeared to attenuate IPAH-PASMC growth at an early time (24 hours) and lasted for at least 72 hours [Figure 4d]. These data indicated that upregulated STIM2 was necessary for the enhanced proliferation of IPAH-PASMC. Inhibition of STIM2 was an effective method to attenuate excessive proliferation of IPAH-PASMC; STIM2 could be a potential target for developing a novel therapeutic approach for patients with IPAH.
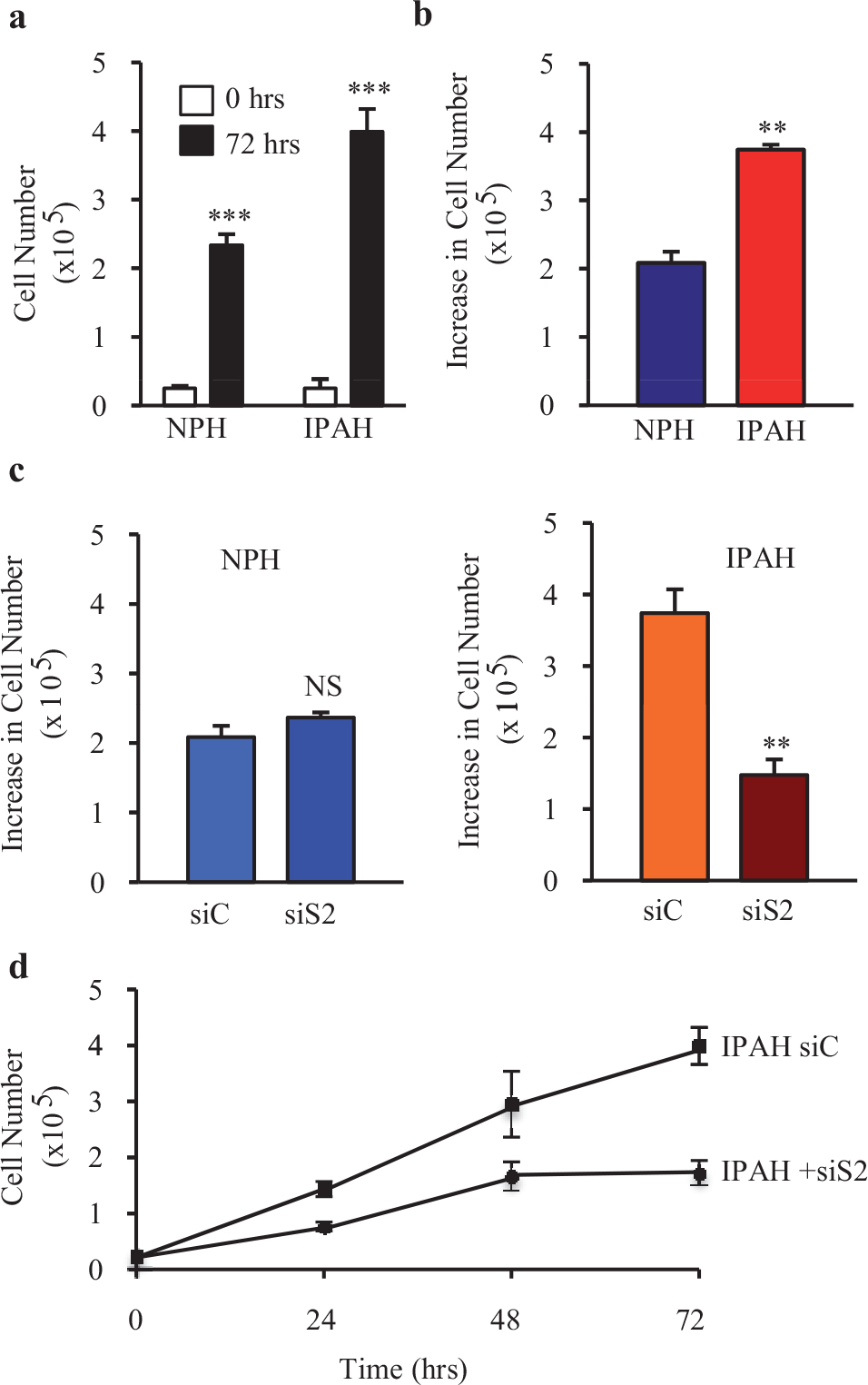
Knockdown of STIM2 mitigates enhanced proliferation in IPAH patients' PASMC. (a) Summary data (mean±SE) showing cell numbers for NPH-PASMC and IPAH-PASMC before (0 hour) and after 72 hours of culture in the growth media. *** P<0.001 versus 0 hour. (b) Summary data showing the increase in cell numbers after 72 hours in NPH-PASMC (blue) and IPAH-PASMC (red). ** P<0.01 versus NPH. IPAH patients' PASMC showed a significantly greater increase in cell number after 72 hours compared to NPH-PASMC. (c) Summary data showing the increase in cell number after 72 hours in NPH-PASMC (left panel) and IPAH-PASMC (right panel) treated with either scrambled siRNA (siC) or siRNA against STIM2 (siS2). Decreasing the protein expression level of STIM2 did not affect the increase in cell number after 72 hours in NPH-PASMC (left panel), but significantly inhibited the increase in cell number after 72 hours in IPAH-PASMC (right panel). ** P<0.01 versus siC. (d) Summary data showing the total number of IPAH-PASMC cultured in growth media at 0, 24, 48, and 72 hours. The cells were treated with scrambled siRNA (square symbols) or siRNA against STIM2 (circle symbols). IPAH-PASMC treated with siRNA-STIM2 had a slower rate of proliferation than IPAH-PASMC treated with scrambled siRNA. The growth curves in IPAH-PASMC treated with scrambled siRNA and siRNA against STIM2 were significantly different
Overexpression of STIM2 is not sufficient to enhance SOCE in normal PASMC
In order to further examine the functional role of STIM2 in IPAH-PASMC, STIM2 was transiently transfected into NPH-PASMC, to determine whether overexpression of STIM2 was sufficient to enhance SOCE and increase proliferation in normal PASMC. STIM2 was overexpressed in NPH-PASMC in a dose-dependent manner [Figure 5]. One microgram of STIM2-cDNA significantly enhanced the protein expression of STIM2 both in HEK-293 cells (data not shown) and NPH-PASMC [Figure 5], and was used to overexpress STIM2 for the experiments described herewith.
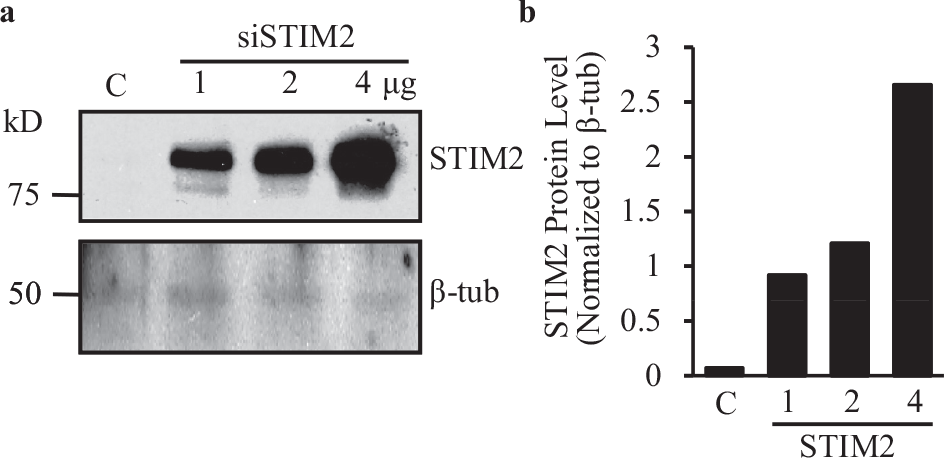
Dose-dependent overexpression of STIM2 in NPH-PASMC transiently transfected with siRNA. (a) Representative Western blot image of STIM2 protein in NPH-PASMC. STIM2 was overexpressed using 1, 2, or 4 μg of DNA. Vector DNA was used as a control (‘C’). β-tubulin (β-tub) was used as a loading control. (b) Quantification of STIM2 protein expression level in NPH-PASMC transfeted with vector DNA (‘C’) and 1, 2, or 4 μg of STIM2-DNA. Values were normalized to β-tubulin. STIM2 protein expression level was increased in a dose-dependent manner in NPH-PASMC transfected with STIM2
Change in [Ca2+]cyt was measured as the ratio of F340 / Foon (F) and normalized to the initial F340 / F380 ratio (F0), as described earlier. SOCE was induced by the passive depletion of SR Ca2+ with 10 μM CPA in NPH-PASMC, superfused with Ca2+-free solution followed by restoration of extracellular Ca2+ (1.8 mM). Overexpression of STIM2 in NPH-PASMC (48 hours after STIM2 transfection) had no significant effect on either the amplitude of the rise in [Ca2+]cyt due to Ca2+ release from the SR [Figure 6b, right panel] or the amplitude of SOCE [Figure 6b, right panel] when compared to the NPH-PASMC transfected with the control vector. In fact, overexpression of STIM2 caused a slight (but not statistically significant) decrease in the peak amplitude of SOCE [Figure 6b, right panel]. These data suggest that overexpression of STIM2 is not sufficient to enhance SOCE in normal PASMC.
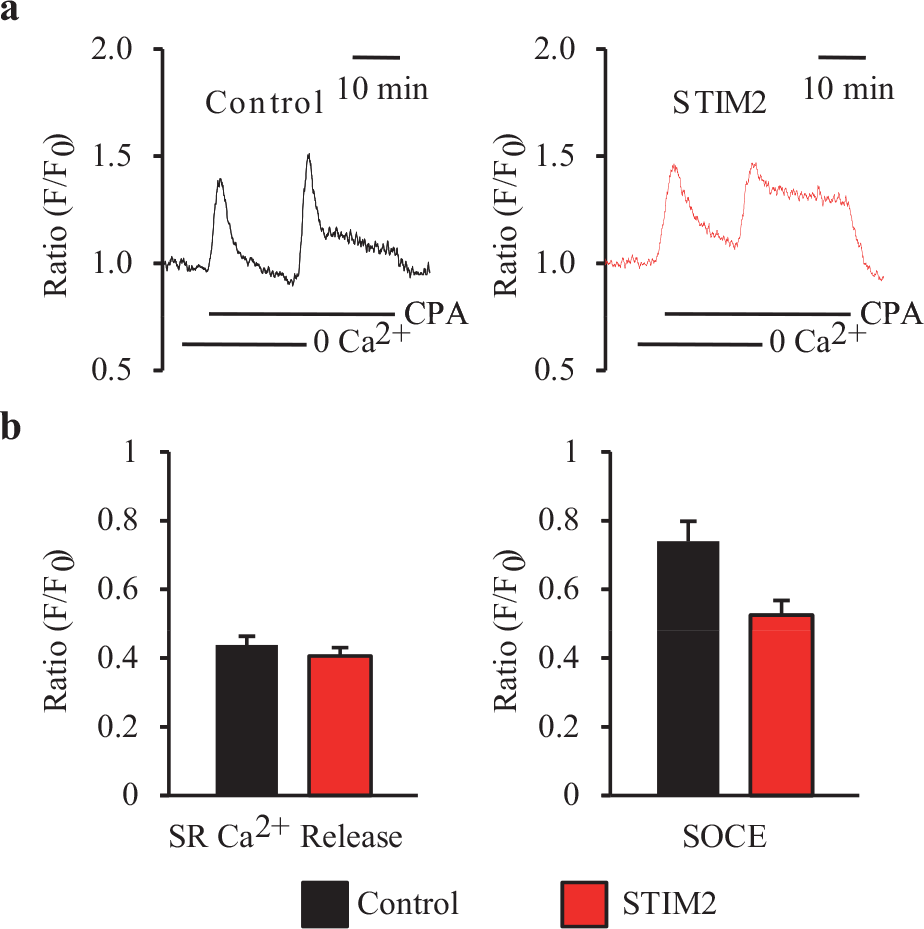
Overexpression of STIM2 is not sufficient to enhance SOCE in normal PASMC. (a) Representative records showing changes in [Ca2+]cyt before, during, and after the application of CPA (10 μM), in the absence or presence of extracellular Ca2+ in the vector control (left panel) and STIM2-transfected (right panel) NPH-PASMC. SOCE was induced by the passive depletion of SR Ca2+, using CPA. (b) Summary data (mean±SE) showing changes in [Ca2+]cyt immediately following the addition of CPA in the absence of extracellular Ca2+, which reflects SR Ca2+ release (SR Ca2+ Release, left panel), in control (black) and STIM2-transfected (red) NPH-PASMC. There was no difference in magnitude of SR Ca2+ release between the two groups. Summary data (mean±SE) showing changes in [Ca2+]cyt immediately following the re-addition of Ca2+ after CPA-induced store depletion (indicative of SOCE, right panel) in control (black) and STIM2-overexpressing PASMC (red). There was no difference in magnitude of SOCE between the two groups
Overexpression of STIM2 is not sufficient to increase proliferation in normal PASMC
In order to examine whether overexpression of STIM2 in normal PASMC would enhance cell proliferation, we measured and compared cell number changes in NPH-PASMC, transfected with a control vector and NPH-PASMC transfected with STIM2. As shown in Figure 7, overexpression of STIM2 failed to enhance the proliferation of NPH-PASMC; the increases in cell number were comparable between the control vector-transfected NPH-PASMC and STIM2-transfected NPH-PASMC. In these experiments, proliferation of PASMC was determined by counting the cells at time 0 and 72 hours. Transfection of PASMC with either STIM2 cDNA or the control vector was performed 48 hours prior to time 0. At time 0, PASMC transfected with STIM2 or the control vector had the same number of cells to start with. After 72 hours, both the control vector- and STIM2-transfected PASMC proliferated, but the increase in cell number was similar between NPH-PASMC transfected with a control vector and STIM2 [Figure 7]. These data suggest that overexpression of STIM2 is not sufficient to enhance the proliferation of normal PASMC.
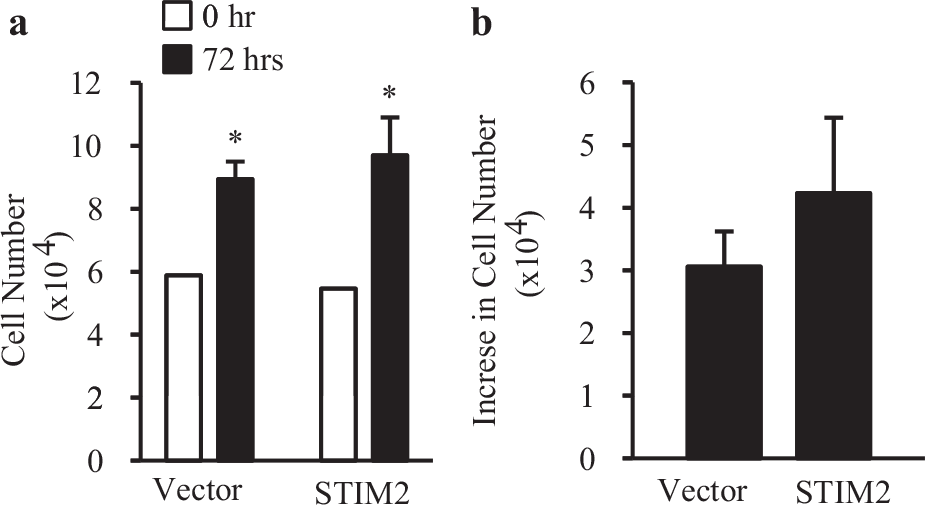
Overexpression of STIM2 does not increase NPH-PASMC proliferation. (a) Summary data for NPH-PASMC proliferation after 72 hours. Cell number was counted at time zero (0 hours: white bar) and 72 hours (72 hours: black bar) and shown as the multiple of 104. After 72 hours, both vector control and STIM2-overexpressing NPH-PASMC increased in cell number. *P<0.05 versus open bars (0 hour). (b) Summary data (mean±SE) showing the increase in cell number after 72 hours in NPH-PASMC. STIM2-overexpressing PASMC (STIM2) did not show a greater increase in cell number after 72 hours compared to control (vector). Cell counts were repeated four times (n=4)
Chronic hypoxia increases protein expression of Orai2 and STIM2 in rat PASMC
Chronic hypoxia causes pulmonary hypertension by increasing PASMC proliferation, migration, and contraction through, at least partially, a Ca2+-dependent mechanism.[25,26] Chronic hypoxia has been reported to inhibit voltage-gated K+ (KV) channels, cause membrane depolarization, open voltage-gated Ca2+ channels, and increase [Ca2+]cyt in PASMC by increased Ca2+ influx through voltage-gated Ca2+ channels. Chronic hypoxia has also been shown to upregulate the TRPC channels in rat PASMC and increase [Ca2+] via enhanced receptor-operated and store-operated Ca2+ influx.[4,5,27–29] To investigate whether the upregulation of STIM2 is also involved in hypoxia-mediated pulmonary vasoconstriction and vascular remodeling, we exposed rat PASMC to hypoxia (PO2=22 mmHg for 48 hours) and examined the protein expression level of STIM2 and Orai2; in these experiments, we used β-tubulin as the loading control. As shown in Figure 8, chronic hypoxia significantly upregulated Orai2 [Figure 8a] and STIM2 [Figure 8b] protein expression levels in normal rat PASMC. These data are consistent with our earlier findings, where STIM2 protein expression levels were increased in PASMC from IPAH patients. Furthermore, increased protein expression of Orai2 in hypoxia-treated rat PASMC might also explain our finding that STIM2 alone is insufficient to enhance SOCE and proliferation in normal PASMC.
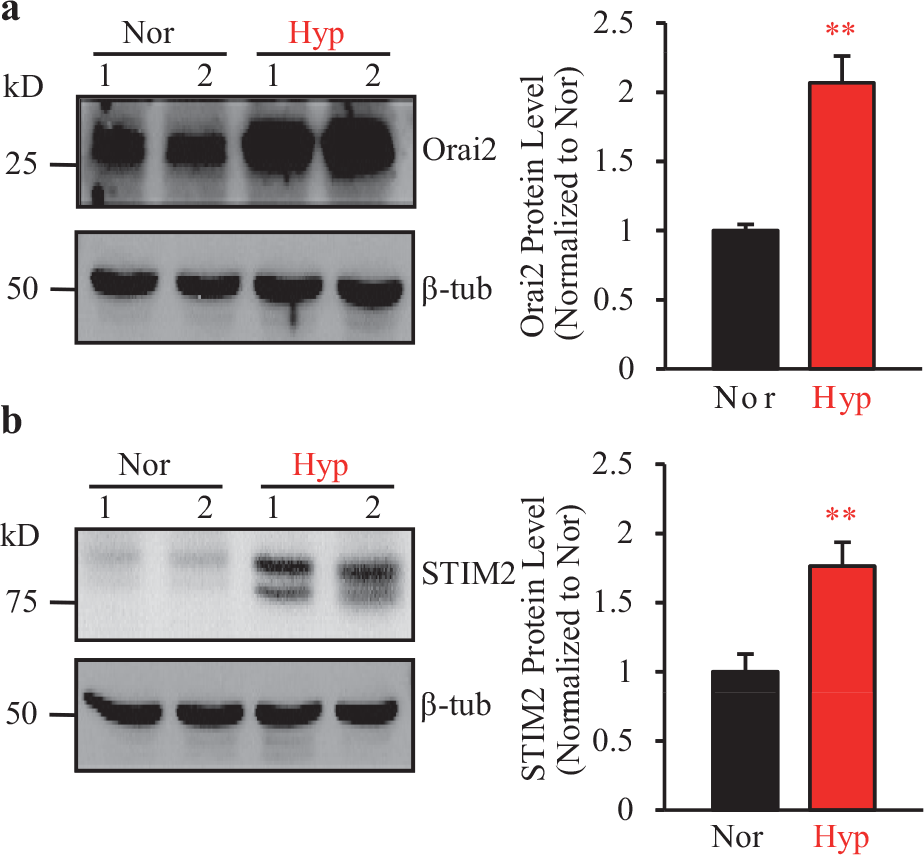
Hypoxia increases protein expression of STIM2 and Orai2 in rat PASMC. (a) and (b). Representative Western blot images (left panels) and summarized data (mean±SE) showing protein levels (right panels) for Orai2 (a) and STIM2 (b) protein in rat PASMC exposed to normoxia (Nor, room air supplemented with 5% CO2) or hypoxia (Hyp, 3% O2 and 5% CO2 balanced in N2 for 48 hours). β-tubulin (β-tub) was used as a loading control. Values were normalized to β-tubulin. Hypoxia increased the protein expression of Orai2 and STIM2 in rat PASMC. ** P< 0.01 versus Nor
Upregulated Orai2 protein expression in PASMC from IPAH patients
As shown in Figure 3 and our previously published data,[17,22,24,30] PASMC from IPAH patients showed enhanced SOCE compared to control PASMC. In addition, IPAH-PASMC showed elevated protein expression of STIM2, but not STIM1 [Figure 1]. Furthermore, while increased protein expression levels of STIM2 were necessary for enhanced SOCE in IPAH-PASMC, overexpression of STIM2 alone was not sufficient to enhance SOCE in NPH-PASMC. Therefore, we examined whether the protein expression of Orai2, a store-operated Ca2+ channel protein, was increased in PASMC from IPAH patients. As shown in Figure 9, the Orai2 protein expression level was significantly higher in IPAH-PASMC than in NPH-PASMC. Taken together, these data suggest that upregulated Orai2, in addition to upregulated STIM2 (this study) and TRPC3 / 6,[22,24] may also be important in enhancing SOCE in PASMC from IPAH patients.
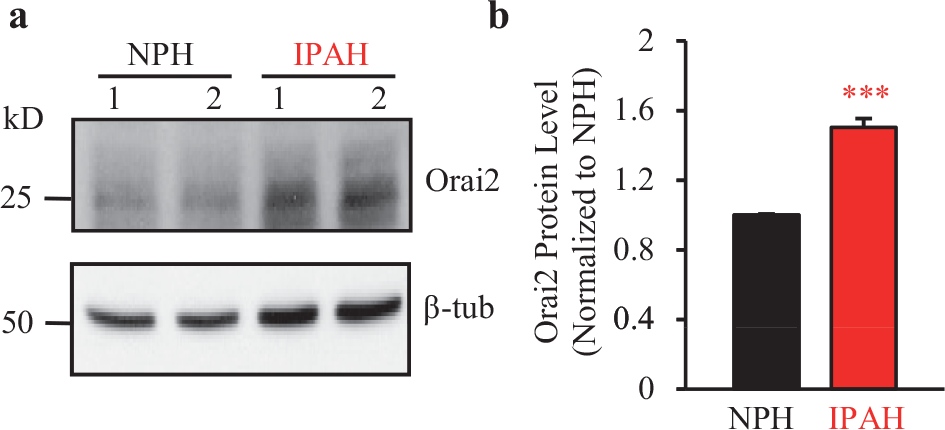
Protein expression of Orai2 was increased in IPAH patients' PASMC. (a) Representative Western blot image for Orai2 protein from IPAH-PASMC and NPH-PASMC. β-tubulin (β-tub) was used as a loading control. (b) Summarized data (mean±SE) comparing protein expression level of Orai2 in IPAH-PASMC to NPH-PASMC. Values were normalized to β-tubulin. IPAH patients' PASMC had significantly higher protein expression level of Orai2 compared to NPH-PASMC. *** P<0.001 versus NPH
DISCUSSION
A rise in [Ca2+]cyt is a major trigger for pulmonary vasoconstriction and is an important stimulus for PASMC proliferation, which leads to pulmonary vascular remodeling.[31,32] Human and animal PASMC functionally express many signal transduction proteins and kinases (e.g., calmodulin, CaMK, MAPK, and calcineurin) and transcription factors (e.g., CREB, c-Fos/c-Jun, c-Myc, NFAT NF-κB) that are sensitive to changes in [Ca2+] or Ca2+-calmodulin (CaM).[33] An increase in [Ca2+]cyt can rapidly increase [Ca2+] in the nuclei[6,34,36] and stimulate nuclear events that are related to cell proliferation. In the cell cycle, it has been well-demonstrated that there are at least four steps that are regulated by Ca2+ or Ca2+/CaM: the transition from the G0 to the G1 phase, the transition from the G1 to the S phase, the transition from the S to the M phase, and the whole process in the M phase.[37–40] In isolated pulmonary arterial rings, removal of extracellular Ca2+ abolished high K+-mediated pulmonary vasoconstriction and significantly inhibited agonist-mediated pulmonary vasoconstriction.[31,41,42] In cultured PASMC, reduction of free Ca2+ concentration in the culture medium significantly inhibited cell proliferation in the presence of growth factors, while depletion of intracellularly stored Ca2+ in the sarcoplasmic or endoplasmic reticulum also significantly attenuated smooth muscle cell proliferation.[43–45] In addition, Ca2+ is an important signal in initiating and guiding cell migration.[46–51] These observations indicate that intracellular Ca2+ signaling, regulated by Ca2+ influx through Ca2+-permeable channels; Ca2+ extrusion via Ca2+-Mg2+ ATPase (Ca2+ pumps) in the plasma membrane; Ca2+ release through IP3 / ryanadine receptors; and Ca2+ uptake via the Ca2+-Mg2+ ATPase (SERCA) on the SR / ER membrane, play an important role in the regulation of smooth muscle cell contraction, migration, and proliferation. Significantly, the upregulated expression of Ca2+-permeable channels and enhanced Ca2+ influx through these channels would thus cause sustained vasoconstriction, stimulate smooth muscle cell migration and proliferation, and ultimately cause vascular wall thickening.
IPAH-PASMC demonstrated significantly enhanced SOCE and proliferated faster compared to NPH-PASMC.[22,24,30] In this study, we have shown that STIM2 is necessary but insufficient for the enhancement of SOCE in IPAH-PASMC. Many studies have shown that STIM1 can enhance SOCE when overexpressed in a variety of cell types.[8,10,12,52,53] This lead us to hypothesize that STIM1 protein expression will be upregulated in IPAH-PASMC. Surprisingly STIM2, but not STIM1 protein expression, is increased in IPAH-PASMC compared to NPH-PASMC. These findings are consistent with the observations which show that protein expression of STIM2 is upregulated in rat PASMC subjected to hypoxia (48 hours). These data suggest that STIM2 may be the important isoform for pathophysiological increase in SOCE in PASMC from IPAH patients.
Further examination showed that STIM2 played an important functional role in both augmented SOCE and enhanced proliferation in IPAH-PASMC. Increased protein expression of STIM2 in IPAH-PASMC was necessary for increased SOCE compared to NPH-PASMC. When the STIM2 protein expression level in IPAH-PASMC was decreased to the level similar to NPH-PASMC, IPAH-PASMC no longer exhibited enhanced SOCE or enhanced proliferation. However, overexpression of STIM2 alone could not enhance SOCE or proliferation in NPH-PASMC, suggesting that STIM2 did not act alone in eliciting the pathophysiological changes in PASMC associated with IPAH. Taken together, these findings suggested that STIM2 was necessary, but not sufficient, for the enhanced proliferation and SOCE found in IPAH-PASMC. Functional interaction of STIM2 with STIM1 and / or Orai and TRPC channels might also be important.
As mentioned earlier, STIM2 overexpression had no effect on SOCE or proliferation in NPH-PASMC, while knockdown of STIM2 in IPAH-PASMC decreased both SOCE and the proliferation. Therefore, it seems that STIM2 was functionally active only in IPAH-PASMC and had no or little function in NPH-PASMC. Several explanations could explain how STIM2 was ‘turned on’ in IPAH, but not in NPH-PASMC. STIM2 could be activated by phosphorylation or another form of post-translational modification. It is widely accepted that Rho/Rho-associated protein kinase (ROCK) signaling is involved in Ca2+ sensitization, proliferation, contraction, and migration in PASMC.[54] Researchers have demonstrated that the Rho/ROCK signaling pathway plays a significant role in the pathogenesis of different experimental models of PAH as well as PAH in patients.[55–60] Perhaps the pathogenesis of IPAH involves RhoA / ROCK signaling though STIM2.
A second explanation for how STIM2 is only active in IPAH-PASMC, but not NPH-PASMC, involves store-operated channels (SOC) on the plasma membrane. Strong evidence suggests that Orai1 interacts with STIM1 and functions as an SOC. Orai1 was discovered through genome-wide RNAi screening. A mutation in Orai1 in patients with severe combined immune deficiency eliminates the Ca2+ release-activated Ca2+ currents (ICRAC), which can then be reconstituted by expressing the wild-type Orai1.[61,62] Orai1 spans the plasma membrane four times, with both the C- and N-termi, in the cytoplasm. Co-expression of STIM1 with Orai1 in the HEK293 cells results in large increases in SOCE and ICRAC compared to the vector control cells.[12,13] The co-immunoprecipitation data shows that STIM1 and Orai1 interact and store depletion significantly increases the amount to an interaction.[63] Furthermore, other isoforms of Orai (e.g., Orai2 and Orai3) are also believed to play a role in SOCE.[62] Our laboratory has previously demonstrated that TRPC channels also function as SOC and are important in PASMC physiology and pathophysiology.[22–2431,64] TRPC channels are important in the regulation of vascular tone, as they regulate the Ca2+ influx required for agonist-induced vasoconstriction and mitogen-mediated smooth muscle cell proliferation. Moreover, the ability of the TRPC channels to alter [Ca2+]cyt without a change in the membrane potential, lends them the ability to modulate vasoconstriction and vasorelaxation through a voltage-independent mechanism. Agonist- and hypoxia-induced pulmonary vasoconstriction is believed to be, at least in part, mediated through the Ca2+ influx, through the TRPC1 and TRPC6 channels.[23,65] Upregulated TRPC channel expression, enhanced SOCE, and increased [Ca2+]cyt are associated with the enhanced proliferation of PASMC isolated from IPAH patients.[21,22] Therefore, increased SOCE and proliferation in the IPAH-PASMC may be functions of both increased protein expression of STIM2 and increased protein expression of SOC, such as, Orai1/2 or TRPC1/3/6, or TRPC in the plasma membrane. Corroborating this idea, we found enhanced protein expression of Orai2 in both the hypoxia-treated rat PASMC and IPAH-PASMC. It is, however, unknown whether STIM2 can functionally interact with both the Orai2 and TRPC channels, to enhance SOCE in PASMC from IPAH patients; this is a research project we are currently pursuing.
In conclusion, upregulated protein expression of STIM2, a Ca2+ sensor that senses the level of Ca2+ in the sarcoplasmic reticulum and mediates SOCE by recruiting and activating SOC in the plasma membrane, is necessary for the augmented SOCE and enhanced proliferation in PASMC from IPAH patients. Downregulation of STIM2 protein expression and / or inhibition of STIM2 protein function in PASMC may be an important target for developing therapeutic approaches for IPAH. As upregulation of STIM2 alone, in normal PASMC, is not sufficient to enhance SOCE, it is likely that STIM2 functionally and physically interacts with other proteins (e.g., Orai2, TRPC3 / TRPC6) involved in forming SOC to regulate SOCE in normal PASMC and cause enhanced SOCE and proliferation in PASMC from IPAH patients.
Footnotes
ACKNOWLEDGMENTS
We thank Amy Zeifman for her critical review of the manuscript, and Anthony Ngo and Dr. Ling Zhu for their technical assistance.
This study has been supported, in part, by grants from the National Institutes of Health (HL066012 and HL 098053 to JX-JY and DK 083506 to AM). M.Y. Song is supported by a pre-doctoral training grant from the National Institutes of Health (T32 DK07202).