
Abstract
Pulmonary hypertension (PH), a progressive disorder associated with significant morbidity and mortality, is caused by complex pathways that culminate in structural and functional alterations of the pulmonary circulation and increases in pulmonary vascular resistance and pressure. Diverse genetic, pathological, or environmental triggers stimulate PH pathogenesis culminating in vasoconstriction, cell proliferation, vascular remodeling, and thrombosis. We conducted a thorough literature review by performing MEDLINE searches via PubMed to identify articles pertaining to PPARΓ as a therapeutic target for the treatment of PH. This review examines basic and preclinical studies that explore PPARΓ and its ability to regulate PH pathogenesis. Despite the current therapies that target specific pathways in PH pathogenesis, including prostacyclin derivatives, endothelin-receptor antagonists, and phosphodiesterase type 5 inhibitors, morbidity and mortality related to PH remain unacceptably high, indicating the need for novel therapeutic approaches. Consequently, therapeutic targets that simultaneously regulate multiple pathways involved in PH pathogenesis have gained attention. This review focuses on peroxisome proliferator-activated receptor gamma (PPARΓ), a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors. While the PPARγ receptor is best known as a master regulator of lipid and glucose metabolism, a growing body of literature demonstrates that activation of PPARγ exerts antiproliferative, antithrombotic, and vasodilatory effects on the vasculature, suggesting its potential efficacy as a PH therapeutic target.
INTRODUCTION
Pulmonary hypertension (PH) is a rare and progressive disorder with a prevalence of 15 cases per million.[1] The hemodynamic definition of PH requires elevation of the mean pulmonary artery pressure >25 mm Hg at rest or 30 mm Hg with exercise and a mean pulmonary-capillary wedge pressure or left ventricular end-diastolic pressure ≤15 mm Hg. The World Health Organization (WHO) now classifies PH into five groups with similar disease mechanisms, histopathologic features and responses to treatment.[2] Current concepts suggest that PH pathogenesis involves three primary processes: vasoconstriction, cellular proliferation/vascular remodeling, and thrombosis.[3] Evolving evidence suggests that peroxisome proliferator-activated receptor gamma (PPARγ), a member of the nuclear hormone receptor superfamily of ligand-activated transcription factors, can favorably modulate cellular proliferation, vascular tone and coagulation. This review provides an overview of PPARγ and considers how PPARγ impacts these primary processes involved in PH pathogenesis.
A thorough literature review was conducted to identify articles pertaining to PPAR as a therapeutic target for the treatment of PH. We performed MEDLINE searches through PubMed in the National Library of Medicine to identify relevant articles. Articles accepted for review included full text journal articles, clinical trials, reviews, guidelines, and randomized controlled trials. Articles not accepted for review included letters, editorials and correspondence, and manuscripts published before 1985.
PPARγ AND PH
Overview of PPARγ biology
Peroxisome proliferator-activated receptors (PPARs) belong to subfamily 1 of the nuclear hormone receptor superfamily (Nuclear Receptors Nomenclature Committee, 1999). There are three distinct isotypes of PPARs (α, β/δ, and γ) that are expressed throughout the body and possess similar structural and functional features. PPARs are responsive to the lipid status of the cell and act as ligand-activated transcription factors. PPAR isotypes are distinguished by their tissue distribution, ligand specificity and target genes. PPARa is most highly expressed in tissues with high catabolic rates such as the liver, kidney, heart, brown adipose tissue, and the intestine.[4,5] PPARβ/δ has the broadest expression pattern with the expression in many cases dependent on the degree of cell proliferation and differentiation.[4,6,7] PPARΓ exists as two isoforms that differ in their N terminus. PPARΓ2 is found in adipose and PPARΓ1 is more widely expressed in tissues such as the brain, vascular tissues, small intestine, and lymphatic tissues.[4,8] Each PPAR isotype is preferentially activated by a wide range of naturally occurring or metabolized lipids derived from the diet or from intracellular signaling pathways, which include n-3 and n-6 family polyunsaturated fatty acids (PUFAs) and eicosanoid products of cyclooxygenase and lipoxygenase.
As illustrated in Figure 1, transcriptional regulation by PPARs requires the formation of heterodimers with retinoid X receptor (RXR) isotypes. PPARs typically form heterodimers with RXRalpha (RXRα). PPAR:RXR heterodimers bind to DNA at sites composed of the hexameric direct repeat sequence, AGGTCA, separated by a single nucleotide. Gene regulation involves ligand-induced conformational changes in the PPAR receptor which mediate interaction with specific coactivator (e.g. steroid receptor coactivator-1 and p300) and corepressor molecules. Coactivator proteins possess either histone acetyltransferase activity or recruit other proteins with this activity to the transcription start site. Acetylation of histone proteins alters chromatin structure, facilitating the binding of RNA polymerase and the initiation of transcription.[9] PPARΓ can also repress gene expression by interfering with the clearance of corepressors from selected promoters.[10] Due to their numerous metabolic and therapeutic actions, PPARs have become pharmacological targets. For example, PPARα agonists such as fibrate medications are used in the treatment of dyslipidemia and may have anti-inflammatory effects. PPARβ/α selective agonists such as GW0742 augment high density lipoprotein (HDL) cholesterol and have been implicated in promoting tissue repair. [9–12] On the other hand, PPARΓ agonists such as rosiglitazone and pioglitazone, used to treat type 2 diabetes, have beneficial effects in vascular disease in animal models.[11,12] These reports emphasize that ligands for PPAR receptors can exert diverse biological effects in numerous organs and tissues. Among these PPAR receptors, PPARΓ is of particular interest in the pulmonary circulation.
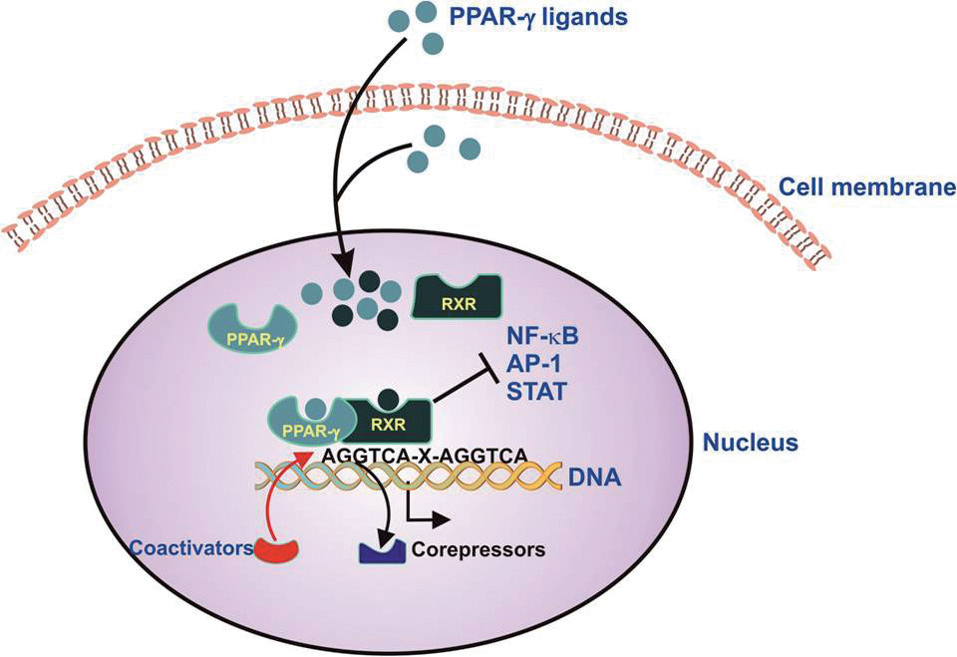
Schematic illustration of PPARγ-mediated gene regulation. PPARγ ligands originating from outside or inside the cell bind to the PPARγ receptor, stimulating formation of a heterodimer with the retinoid X receptor (RXR). The PPARγ-RXR heterodimer binds to PPAR response elements (PPRE) comprising hexameric repeats of the sequence, AGGTCA-X-AGGTCA. This active heterodimer recruits coactivators to and/or derecruits corepressor molecules from the transcriptional start site, leading to increased transcription of selected genes. Activation of PPARγ can also inhibit the expression of genes regulated by specific proinflammatory transcription factors such as NF-kB, AP-1, and STAT
The role of altered PPARγ expression in PH
PPARγ is abundantly expressed in many cell types in the lung, including those of the pulmonary vascular wall such as endothelial and smooth muscle cells. Several studies indicate that expression of PPARγ is reduced by conditions associated with PH. Ameshima and colleagues were the first to demonstrate that compared to normal or chronic obstructive pulmonary disease (COPD) patients, patients with PH had significantly reduced or absent expression of PPARγ in precapillary arteriolar plexiform lesions.[13] PPARγ expression was markedly reduced in endothelial cells isolated from PH patients when compared to normal patients (unpublished observation). These clinical findings have also been corroborated using in vivo experimental models of PH. For example, PPARγ expression was reduced in pulmonary vascular lesions in the rat model of hypoxia-induced PH.[13,14] Similarly, using in vitro cell culture models, increased shear stress or hypoxia was demonstrated to directly alter PPARΓ expression. Exposure of ECV304 endothelial cells to increased fluid shear stress decreased PPARγ expression.[13] Similarly, exposure of endothelial cells to 1% hypoxia decreased expression of PPARγ.[15] Collectively, these findings suggest that PPARγ expression is reduced in PH and that cells exposed to conditions that promote PH have decreased PPARγ expression. These reductions in PPARγ could contribute to an abnormal, proliferative, and apoptosis-resistant endothelial cell phenotype.
To further examine the role of PPARγ in pulmonary vascular biology, more recent studies have employed PPARγ knockout mice. Because global deletion of PPARγ results in embryonic lethality,[16] investigators have examined experimental animals with tissue-targeted deletion of PPARγ. For example, Guignabert and coworkers reported that targeted deletion of PPARγ in the vascular endothelium of mice (ePPARγ−/−) results in spontaneous PH with right ventricular hypertrophy and muscularization of small distal pulmonary arteries.[17] The ePPARγ−/− mice exposed to chronic hypoxia (10% O2) for 3 weeks developed a similar degree of PH as wild-type control mice. However, following cessation of hypoxia, PH persisted longer in the ePPARγ−/− mice compared to wild-type mice exposed to hypoxia, suggesting that reduced endothelial PPARγ signaling is sufficient to cause mild PH and impair recovery from chronic hypoxia exposure.[17] Targeted deletion of PPARγ from smooth muscle (smPPARγ−/−) also resulted in spontaneous PH in mice.[18] Microarray analysis of bovine pulmonary artery endothelial cells following treatment with a PPARγ antagonist revealed alterations in the expression of numerous genes including those that might stimulate cell cycle progression and proliferation.[19] Taken together, these reports suggest that loss of PPARγ function in pulmonary vascular wall cells stimulates PH pathogenesis.
PPARγ activation ameliorates experimental PH
Mounting experimental evidence indicates that PPARγ stimulation ameliorates PH development in animal models of PH. Monocrotaline (MCT)-induced PH and vascular remodeling in the rat were attenuated by treatment with the PPARγ ligands, pioglitazone or troglitazone.[20] Interestingly, PPARγ ligands also inhibited MCT-induced vascular wall thickening and staining for proliferating cell nuclear antigen, suggesting that PPARγ ligands suppressed cell proliferation and vascular remodeling.[20] In Wistar-Kyoto rats exposed to continuous hypobaric hypoxia for 3 weeks, treatment with rosiglitazone attenuated hypoxia-induced right ventricular hypertrophy and vascular smooth muscle cell (VSMC) proliferation, as well as pulmonary vascular collagen and elastin deposition, infiltration of c-Kit-positive cells into the adventitia, and matrix metalloproteinase-2 (MMP-2) activity. In this study, rosiglitazone failed to attenuate hypoxia-induced increases in pulmonary artery pressure, an observation attributed to the inability of PPARγ ligands to modulate Rho kinase signaling, a critical mediator of pulmonary vasoconstriction.[21] Hansmann and colleagues reported that ApoE knockout mice fed high fat diets developed significant increases in right ventricular systolic pressure, pulmonary vascular remodeling and right ventricular hypertrophy and that administration of PPARγ ligands in this model attenuated PH.[22] An elegant series of experiments in this model provided evidence that PPARγ ligands attenuated PH by inhibiting platelet derived growth factor (PDGF) signaling.
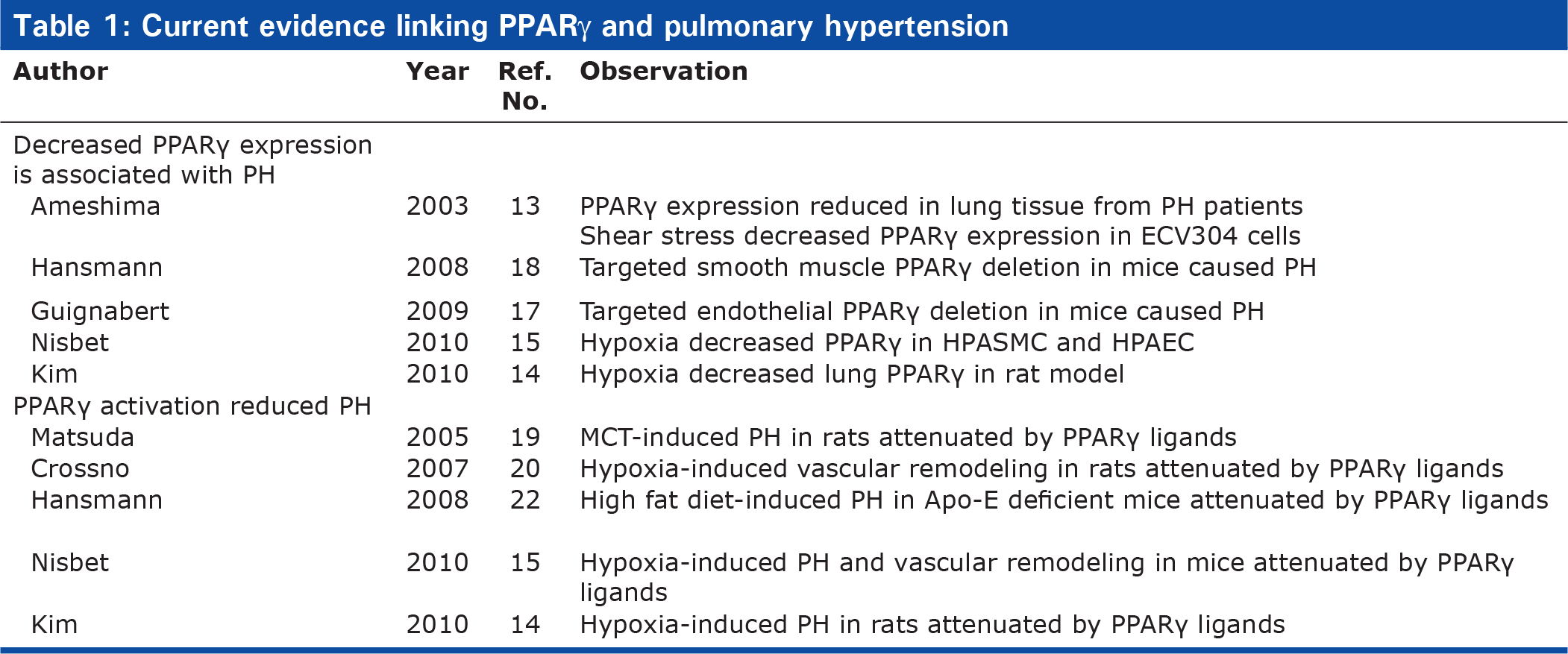
Male C57Bl/6 mice exposed to chronic hypoxia (10% O2) for 3 weeks developed PH that was attenuated by treatment with the PPARγ agonist, rosiglitazone (10 mg/kg/day by gavage) during the final 10 days of hypoxia exposure.[15] Rosiglitazone treatment also reduced hypoxia-induced right ventricular hypertrophy and muscularization of small pulmonary arterioles. From a therapeutic perspective, this study also demonstrated that rosiglitazone could reverse the established PH by introducing rosiglitazone treatment only after animals developed PH.[15] The mechanisms of these therapeutic effects were attributed to PPARγ-mediated reductions in Nox4 expression, oxidative stress, and PDGF signaling in the lung. Collectively, these reports indicate that PPARγ ligands attenuated pulmonary vascular remodeling and hypertension caused by a variety of stimuli in experimental models. The effect of alterations in PPARγ expression and activation on PH in various experimental models is summarized in Table 1. These studies have begun to identify specific pathways modulated by PPARγ in the pathogenesis of PH. Additional evidence, reviewed below, suggests that PPARγ has the potential to regulate a diverse spectrum of pathways and mediators implicated in PH. The ensuing section will consider how targeting PPARγ can potentially modulate additional pathways fundamental to the pathogenesis of PH.
Current evidence linking PPARγ and pulmonary hypertension
ASPECTS OF PH PATHOGENESIS THAT ARE POTENTIALLY REGULATED BY PPARγ
Vascular tone in PH
Common mechanisms in the pathogenesis of PH are endothelial dysfunction, reduced endothelial production of vasodilators, and increased production of vasoconstrictors leading to vasoconstriction and increased pulmonary vascular resistance. Recent advances in PH therapeutics have sought to promote vascular function by restoring vasodilator levels. For example, circulating levels of the vasodilator, prostacyclin, are decreased in PH,[23] and the administration of prostacyclin or its analogues represents a significant advance in PH therapy.[24] Nitric oxide (NO)-mediated vasorelaxation is also impaired in PH.[25] Phosphodiesterase type 5 inhibitors which prolong NO-mediated increases in cGMP are now employed in selected patients with PH.[24] Conversely, levels of vasoconstrictors such as endothelin-1 (ET-1) and thromboxane (TXA2) are increased in PH patients.[26] By blocking these vasoconstrictive effects, both endothelin receptor antagonists and calcium channel blockers comprise additional strategies in the current PH therapeutic armamentarium. In addition to altering vascular tone, these agents may also modulate structural changes in the pulmonary vasculature and vascular remodeling, sequelae of vascular injury and increased intraluminal pressure or flow. While the current therapies target individual mediators or mechanisms in PH pathogenesis, PPARγ may simultaneously modulate several of these pathways involved in PH pathogenesis as described below.
Prostacyclin
As a critical regulator of pulmonary vascular function, the endothelial-derived mediator, prostacyclin, is a potent vasodilator that inhibits platelet aggregation and exerts anti-inflammatory, anti-thrombotic, and anti-proliferative vascular effects.[27] Overexpression of prostacyclin synthase protected mice from chronic hypoxia-induced PH, whereas prostacyclin-receptor deficient mice were sensitized to hypoxia-induced PH.[28] Prostacyclin synthase expression was reduced in the pulmonary arteries of patients with severe PH compared to normal subjects, and the vascular endothelium was found to be the major site of lung vascular prostacyclin synthase expression.[29] In patients with PH, prostacyclin derivatives decreased urinary isoprostane metabolites, an index of oxidative stress, without altering TXA2.[30] Currently, augmenting prostacyclin levels constitutes a therapeutic strategy in PH, but the precise cellular mechanisms responsible for prostacyclin-mediated benefits remain to be defined. The classical signaling pathway activated by prostacyclin involves binding the G-protein coupled cell surface prostacyclin receptor (IP), which when activated, stimulates adenyl cyclase and increases cellular cAMP content. However, prostacyclin and its analogues can also activate PPAR receptors including PPARS and PPARγ to mediate biological effects.[31,32] Activation of the PPARγ receptor with thiazolidinedione also reduced systemic vascular production of the potent vasoconstrictor, thromboxane,[33] and attenuated iNOS and Cox-2 upregulation.[34] These reports emphasize that PPARγ can regulate prostanoid production and that additional studies will be required to determine if PPARγ can regulate prostacyclin and its metabolites in the pulmonary circulation.
Nitric oxide
In addition to prostacyclin, NO represents an additional endothelium-derived vasodilator whose bioavailability is reduced in PH.[35,36] Downregulation of the enzyme that produces nitric oxide, endothelial nitric oxide synthase (eNOS), has been described in PH in some studies,[37] whereas others report unchanged or increased levels of the enzyme. (More consistent evidence demonstrates that endothelium-derived, NO-mediated vasodilation is impaired in models of PH.) Thus, it is likely that reductions in NO bioavailability in PH are more closely related to post-translational alterations in eNOS regulation and/ or enhanced NO degradation rather than reduced eNOS expression. The critical role of NO bioavailability in PH is supported by the evidence that genetic deletion of eNOS enhanced susceptibility to hypoxia-induced PH,[38] a defect reversed by adenoviral-mediated transfection of the pulmonary vasculature with eNOS.[39] Furthermore, overexpression of eNOS attenuated hypoxia-induced PH.[40] In addition, NO inhalation improves pulmonary hemodynamics and quality of life in a subset of patients with PH,[41] and recent advances in cell-based eNOS gene transfer to the lung have demonstrated that eNOS can reverse the established PH in animal models and facilitate restoration of the pulmonary microvasculature.[42]
NO is produced constitutively in vascular endothelial cells from the amino acid, L-arginine, by the Type III, eNOS isoform. Enzyme activity is largely regulated by intracellular Ca2+, cofactor availability, and eNOS post-translational modifications including phosphorylation and interaction with other proteins such as caveolin and heat shock protein 90 (hsp90).[43] Derangements in eNOS phosphorylation and interactions between eNOS and caveolin have been reported in PH. PPARγ ligands can affect these post-translational pathways.[44,45] PPARγ increased endothelial NO release by reducing the inhibitory interaction between eNOS and caveolin, increasing interactions between eNOS and the molecular chaperone, hsp90, and enhancing phosphorylation of eNOS on serine 1177, all post-translational modifications associated with enhanced eNOS activity.[46,47] PPARγ agonists can increase NO bioavailability by blunting the degradation of inducible nitric oxide synthase (iNOS) and by decreasing serum levels of asymmetric dimethylarginine (ADMA), the endogenous inhibitor of NOS.[48–50]
Once produced, endothelial-derived NO reduces: 1) vascular tone,[51] 2) platelet activation and aggregation,[52] 3) stimulated vascular smooth muscle proliferation.[53] and 4) leukocyte adherence.[54] To exert these biological effects, NO must diffuse into the vascular wall where its activity may be limited by local concentrations of superoxide. Superoxide combines at diffusion-limited rates with NO, forming the potent oxidant, peroxynitrite, thereby reducing the vascular protective effects of NO and enhancing oxidative stress. Peroxynitrite also oxidizes the NOS cofactor, tetrahydrobiopterin.[55] Deficiency of tetrahydrobiopterin alters electron flow through eNOS to molecular oxygen rather than arginine, producing superoxide rather than NO, a condition referred to as eNOS uncoupling. Enhanced superoxide production in the vascular wall may, therefore, reduce NO bioavailability through multiple mechanisms. As a result, NO bioavailability can be regulated not only by the rate of NO formation, but also by the rate of NO degradation. Thus, increased superoxide generation constitutes an important mechanism of NO inactivation and endothelial dysfunction in the vascular wall. Current evidence indicates that PPARγ can stimulate endothelial NO release and simultaneously reduce superoxide generation in vascular endothelial cells, suggesting that PPARγ ligands could enhance NO bioavailability and the vascular protective effects of NO to favorably modulate vasoconstriction and PH.[46,47]
Endothelin-1
The potent vasoconstricting polypeptide, ET-1, has been implicated in PH pathogenesis. ET-1 receptors are upregulated in the lung in both animal models and patients with PH.[25] ET-1, as well as endothelium-derived reactive oxygen species (ROS), attenuated NO-dependent pulmonary vasodilation, following exposure to chronic hypoxia in isolated rat lungs.[56] ET-1-induced pulmonary vasoconstriction was markedly reduced by administration of Cu/Zn superoxide dismutase and was completely attenuated in gp91phox deficient mice.[57] These findings suggest that NADPH oxidase and superoxide play an important role in pulmonary vascular effects of ET-1. ET-1 receptor antagonists have been employed in patients with PH to improve functional status and other indices of PH related morbidity,[56] further suggesting that ET-1 is an important mediator of pulmonary vascular dysregulation. Emerging evidence in several disease states indicates that PPARγ activation attenuates ET-1 signaling. PPAR ligands inhibited ET-1 secretion by vascular endothelial cells in vitro.[57–60] Similarly, in non-diabetic patients with metabolic syndrome, treatment with rosiglitazone reduced several markers of vascular inflammation including plasma levels of ET-1 and improved markers of metabolic control, while lowering blood pressure and improving flow-mediated vasodilation.[61] PPARγ activation also reduced hypertrophy and anti-apoptotic effects caused by ET-1 in cardiac myocytes in vitro through altered nuclear factor of activated T cells (NFAT) signaling. Treatment with PPARγ ligands in several rat models of hypertension reduced ET-1 expression in cardiac and vascular tissues.[62] Collectively, these findings suggest that PPARγ activation can attenuate expression of ET-1 in cardiovascular tissues in response to a variety of stimuli and can attenuate ET-1-mediated signaling in selected models. Potential pathways by which PPARγ regulates vascular tone are illustrated in Figure 2.
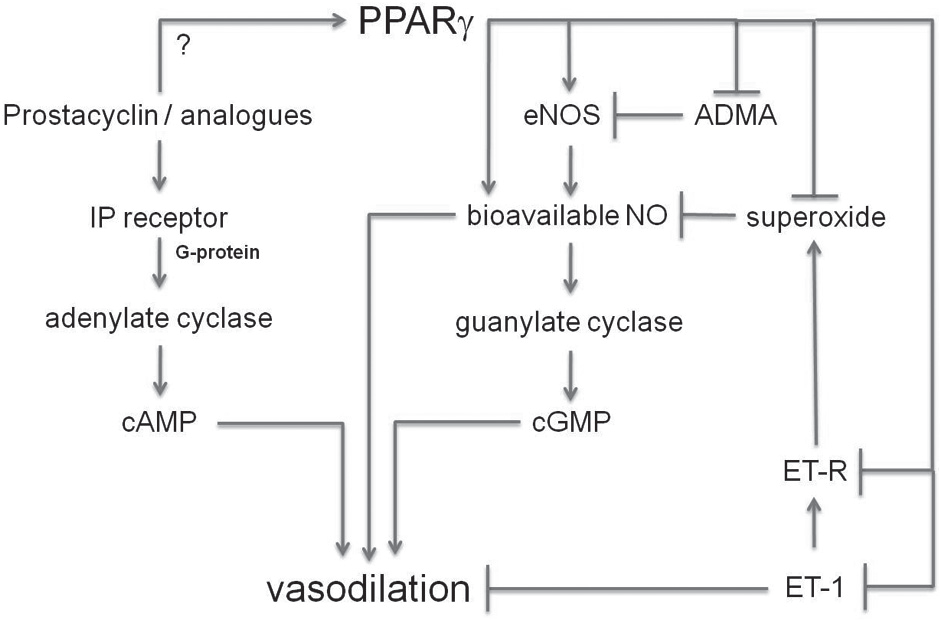
Potential pathways by which PPARγ regulates vascular tone. Activation of PPARγ potentially modulates several pathways involved in the regulation of vascular tone. In addition, prostacyclin or its analogues may activate PPARγ. Arrowheads at the end of lines denote stimulatory effects, whereas perpendicular lines denote inhibitory effects. (ADMA – Asymmetric dimethyl arginine; cAMP – Cyclic adenosine nucleotide monophosphate; cGMP – Cyclic guanosine nucleotide monophosphate; eNOS – Endothelial nitric oxide synthase; ET-1 – Endothelin-1; ET-R – Endothelin-1 receptor; NO – Nitric oxide; IP receptor – Prostacyclin receptor)
Abnormal vascular remodeling, inflammation, and cell proliferation in PH
Remodeling of the pulmonary vasculature can reduce its cross-sectional diameter and compliance, increase pulmonary vascular resistance, and contribute to sustained PH. Studies examining the molecular mechanisms underlying pulmonary vascular remodeling have implicated growth factor pathways as well as matrix remodeling in the development and progression of PH.[63] Inflammation also plays a significant role in altered pulmonary vascular function during the development of PH.[64] Inflammatory markers are elevated in PH, and the plexiform lesions that characterize severe PH are surrounded by macrophages, T and B lymphocytes, and dendritic cells.[65] These cells may exacerbate PH by releasing growth factors, ROS and additional cytokines.[66] Chemokines such as CX3CL1, CCL5 and MCP-1 which recruit inflammatory cells are elevated in PH patients.[67–69] Therefore, agents that target the generation of these oxidative and inflammatory stimuli in the pulmonary vascular wall may reduce vascular dysfunction and attenuate the development or progression of PH. Relationships between PPARγ and these complex pathways implicated in PH pathogenesis are illustrated in Figure 3.
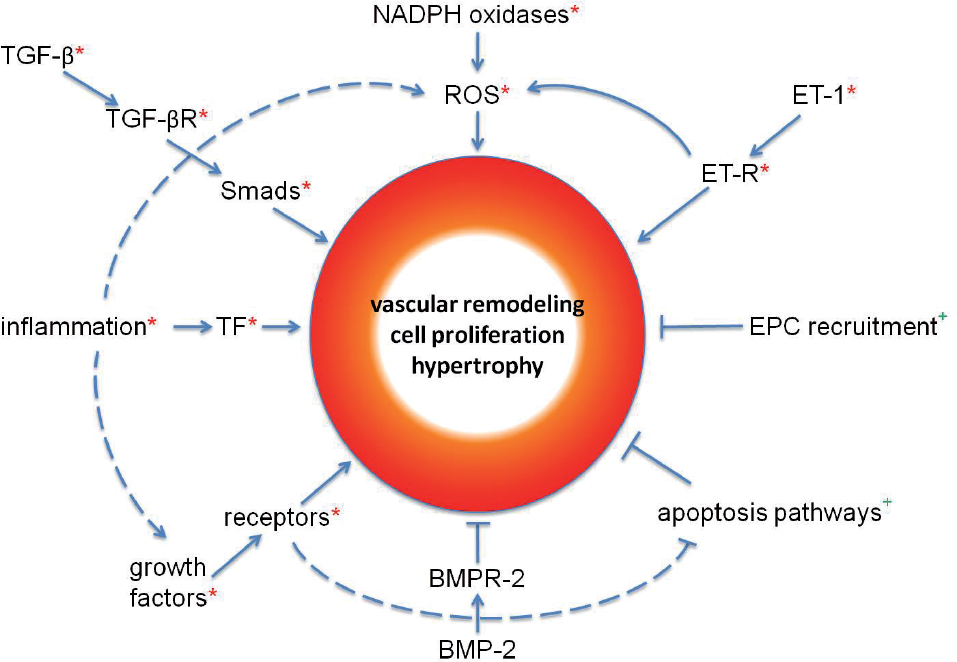
Potential pathways by which PPARγ regulates vascular remodeling, cell proliferation and hypertrophy in PH. PPARγ has the potential to regulate a complex variety of pathways that are involved in remodeling of the pulmonary vasculature during the development of PH. Arrowheads at the end of lines denote stimulatory effects, whereas perpendicular lines denote inhibitory effects. *Pathway components that are inhibited by PPARγ, +pathway components that are stimulated by PPARγ. (BMP-2 – Bone morphogenetic protein-2; BMPR-2 – BMP-2 receptor; EPC – endothelial progenitor cell; ET-1 – Endothelin – 1; ET-R – Endothelin-1 receptor; ROS – Reactive oxygen species; TF – Inflammatory transcription factors (e.g. NF-kB); TGF-β – Transforming growth factor beta; TGFβR – TGF-β receptor)
NADPH oxidases
NADPH oxidases, a major source of superoxide production in the vasculature, have been implicated in PH and contribute to endothelial dysfunction and vascular cell proliferation.[70,71] Originally described in phagocytic cells, the gp91phox-based NADPH oxidase is a multicomponent, membrane-associated, enzyme that catalyzes the one electron reduction of oxygen to superoxide, using NADH or NADPH as the electron donor.[70] The classical phagocytic NADPH oxidase is composed of several components or subunits including the membrane-bound gp91phox (Nox2) and p22phox subunits as well as the cytosolic p47phox and p67phox subunits that, when stimulated, combine with the small G-protein, rac, and translocate to the membrane to activate the enzyme complex. On the other hand, in nonphagocytic cells, the catalytic moiety of NADPH oxidases is composed of one or more Nox2 homologues, Nox 1, 3, 4, 5, Duoxl or Duox2.[72] These Nox homologues associate with the membrane-bound p22phox subunit and are differentially regulated and targeted to distinct subcellular loci, suggesting that these oxidases serve unique roles in cell function. Nox1 and 3 are activated through interactions with rac and the p47phox and p67phox homologues, NOXA1 and NOXO1.
Current evidence indicates that Nox4 expression is increased in hypoxia-induced PH in the mouse and in the pulmonary vasculature of patients with PH.[15,73] Nox4 is highly expressed in vascular wall cells including smooth muscle and endothelial cells where it is constitutively active.[74] Furthermore, hypoxia increased Nox4 expression and pulmonary artery smooth muscle cell (PASMC) proliferation,[73] and Nox4 stimulated the proliferation of endothelial and smooth muscle cells.[75] Hypoxia stimulated Nox4 expression and cell proliferation in the mouse lung in vivo and in human pulmonary artery endothelial cell (HPAEC) and human pulmonary artery smooth muscle cell (HPASMC) in vitro.[15] Treatment with rosiglitazone during the last 10 days of hypoxia exposure reduced Nox4 levels and ROS production and attenuated hypoxia-induced PH, right ventricular hypertrophy and vascular remodeling. Similarly, rosiglitazone attenuated hypoxia-induced Nox4 expression and proliferation in human PAEC and PASMC in vitro. Taken together, these findings suggest that NADPH oxidases are important mediators of cell proliferation and vasoconstriction in PH that can be regulated by PPARγ [summarized in Figure 4].
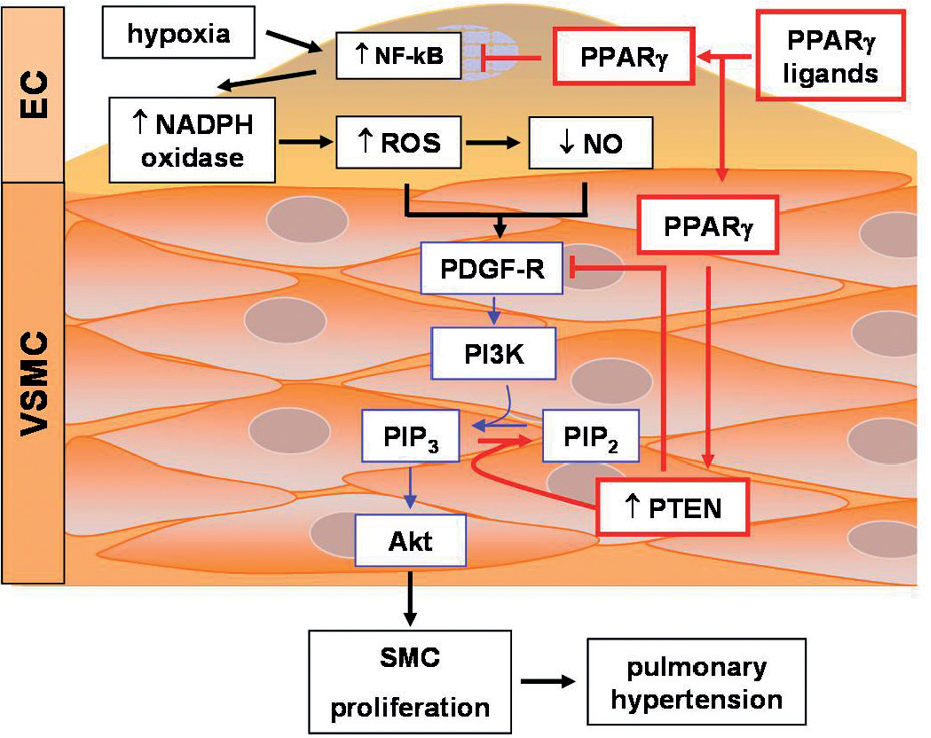
The effects of PPARγ activation on chronic hypoxia-induced PH and PDGF signaling. PPARγ activation decreases oxidative stress and hypoxic vasoconstriction by blunting hypoxia-induced increases NADPH oxidase expression and decreases in NO, respectively. PPARγ stimulated PTEN expression blocks PDGF-mediated vascular smooth muscle cell proliferation. Arrowheads at the end of lines denote stimulatory effects, whereas perpendicular lines denote inhibitory effects
Although increases in Nox4 mRNA levels were associated with increased Nox4 activity,[76] detailed understanding of Nox4 transcriptional regulation remains to be established. Nox4 induction has been reported in response to diverse stimuli including hypoxia in kidney and ischemia in brain.[71] In smooth muscle cells, activators of Nox4 transcription include urokinase, plasminogen activator, angiotensin II, transforming growth factor-beta 1 (TGF)-β1, and tumor necrosis factor (TNF)-α.[77] In contrast, in endothelial cells, oscillatory shear stress[78] and PPARγ activation[79] suppressed Nox4 mRNA levels. However, few studies have examined regulatory elements in the Nox4 promoter. Hypoxia stimulated activation of the Nox4 promoter in part through nuclear factor (NF)-KB-mediated signaling and enhanced p65 binding to the Nox4 promoter which increased Nox4 expression and activity.[80] Furthermore, treatment with rosiglitazone inhibited hypoxia-induced Nox4 expression and activity, proliferation, and p65-Nox4 promoter interaction.[80] HIF-1α and the E2F family transcription factors also activated the Nox4 promoter although their regulation by PPARγ at this site has yet to be confirmed.[81] Based on evidence that Nox4 stimulates smooth muscle and endothelial cell proliferation, these findings suggest that activation of PPARγ can attenuate the proliferation of pulmonary vascular wall cells that may participate in the pathogenesis of PH.[82] Coupled with the ability of NADPH oxidases to attenuate NO bioavailability, Nox4, in particular, and NADPH oxidases, in general, may be an important target of PPARγ amelioration of PH.
PDGF signaling
PDGF participates in PH pathogenesis. Two genes (A and B) produce three biologically active forms of PDGF protein (AA, AB, and BB).[83,84] These proteins activate one or more PDGF receptors (αα, αβ, or ββ) to stimulate cell migration and survival. Ligand binding promotes PDGF receptor tyrosine autophosphorylation and subsequent activation of several downstream signaling pathways including Src, phosphatidylinositol 3 kinase (PI3K), phospholipase Cγ, and Ras. These signaling pathways are largely activated by recruitment of these enzymes to PDGF-R SH-2 domains. The composition of the downstream signaling pathways activated by PDGF and their integration into specific cellular responses continue to be defined.[85] Although the expression of PDGF and its receptors is limited in the vascular wall at baseline, several pathological stimuli, including alterations in blood pressure and shear stress, induce peptide and receptor expression.[84] PDGF receptor expression was increased in the lungs of patients with PH, and the PDGF receptor antagonist, imatinib, reversed MCT- or hypoxia-induced PH in rodents and improved pulmonary vascular resistance and exercise capacity in a patient with severe idiopathic ph.[86–88] PPARγ ligands attenuated hypoxia-induced PDGF activation in a mouse model of PH in vivo.[15] Coupled with reports that NO inhibits PDGF signaling and that PPARγ ligands inhibit PDGF-stimulated VSMC migration in vitro,[89–91] these studies suggest that PPARγ can regulate important proliferative signaling pathways in experimental PH, including those activated by PDGF.
PDGF receptor activity can also be regulated by phosphatase and tensin homologue deleted on chromosome 10 (PTEN), a dual specificity phosphatase that dephosphorylates both lipid and protein substrates.[92] PTEN catalyzes the removal of the phosphate moiety from the 3-position of the phosphatidylinositol ring, converting the second messenger, PI(3,4,5)P3, to PI(4,5)P2.[93] PTEN and PI3K thereby have opposing actions on cellular levels of PI(3,4,5)P3. PTEN also dephosphorylates the PDGF receptor. Because PDGF receptor activation mediates cell proliferation and migration in part through stimulation of PI3K, PTEN can inhibit PDGF signaling both at the receptor and through lowering of PI(3,4,5)P3 levels, thereby lowering the activity of PI3K-related downstream mediators such as the protein kinase B, Akt, which mediates survival, growth, and proliferative signals by inhibiting apoptosis.
PTEN activity is regulated at the transcriptional and post-translational level. At the transcriptional level, pathological stimuli such as ischemia reduced PTEN expression and promoted hypertrophy and remodeling.[94,95] Limited evidence suggests that NF-KB activation may lead to suppression of PTEN expression.[96,97] On the other hand, the PTEN promoter contains two PPAR response elements, and several studies demonstrated that PPARγ ligands stimulated PTEN expression.[98,99] Furthermore, PTE6N overexpression reduced VSMC proliferation and migration and inhibited injury-induced vascular remodeling in vivo.[100,101] PTEN activity is inhibited by ROS which reversibly oxidize cysteine residues in the phosphatase active site.[102,103] In fact, NADPH oxidase-derived ROS facilitated PDGF signaling by inhibiting PTEN.[104] These reports suggested that chronic hypoxia caused sustained PI3K/Akt activation, in part, through the generation of NADPH oxidase-derived ROS that stimulated PDGF and inhibited PTEN signaling pathways. A recent report confirmed that chronic hypoxia increased PDGF receptor activation and reduced PTEN expression in the lung.[15] Furthermore, treatment with rosiglitazone attenuated hypoxia-induced PDGF receptor activation and restored PTEN levels in hypoxic mice to levels comparable to control animals. The ability of PPARγ ligands to simultaneously stimulate PTEN expression and lower oxidative stress-induced PTEN inactivation while attenuating PH provides a unique strategy to lower PDGF receptor phosphorylation and activation and reduce cellular PI(3,4,5)P3 levels. These integrated effects may contribute to the ability of PPARγ to attenuate pulmonary VSMC proliferation and hypertrophy as well as vascular remodeling caused by chronic hypoxia. A hypothetical schema depicting relationships between PPARγ, NADPH oxidase, PDGF, and PTEN is provided in Figure 4.
ALTERED BONE MORPHOGENETIC PROTEIN (BMP) AND TGF-β1 SIGNALING IN PH
BMP and TGF-β1 belong to the TGF beta superfamily of growth factors which are involved in multiple cellular processes including proliferation, differentiation, inflammation, and immunity.[105,106] All TGF-β superfamily ligands are generated as inactive dimeric precursor proteins that are subsequently cleaved by proteases, activated and secreted.[105] The ligands bind to one of two types of serine/threonine kinase receptors (type I and II) causing Smad substrate recruitment, phosphorylation, transduction of intracellular signals and nuclear translocation.[106–108] Approximately 70% of patients with familial pulmonary arterial hypertension and 11-40% with idiopathic pulmonary arterial hypertension (IPAH] have germline mutations in bone morphogenetic protein receptor-2 (BMPR-2), a receptor needed for normal vascular development.[109] BMPR-2 expression is decreased in some PH cases without identified BMPR mutation.[110] In comparison to unaffected patients, IPAH patients have altered cellular growth responses to TGF-β1 and BMP signals which favor VSMC proliferation.[106,111] Specifically, in IPAH patients, TGF-β1 induces a heightened SMC proliferative response and BMP signaling fails to confer an expected antiproliferative and proapoptotic effect.[49] TGF-β1 signaling is responsible for pulmonary artery remodeling in rat models as evidenced by the attenuation of SMC migration and muscularization of small pulmonary arteries that occurred upon the inhibition of the TGF-β1 receptor activin like kinase-5 (ALK-5).[112]
Cell culture experiments reveal that TGF-β1 promotes smooth muscle proliferation via an autocrine induction of PDGF and Nox4.[113] Mechanistically, Nox4-derived ROS may cause transient oxidative inactivation of counterbalancing phosphatases involved in kinase-based cell growth cascades, effectively promoting cell cycle transition and proliferation. Nox4 and TGF-β1 signaling are closely linked and both increase in response to hypoxia and mediate proliferation of HPASMC in vitro.[15,114] Specifically, TGF-β1 was found to be the proximal mediator of HPASMC proliferation through a cascade involving sequential signaling of phosphatidylinositol 3-kinase (PI3K) and serine/threonine kinase (Akt) phosphorylation, insulin-like growth factor binding protein-3 (IGFBP-3), and Nox 4.[114] Dominant negative mutations of the TGF-β type II receptor ameliorated hypoxia-induced pulmonary vascular remodeling in mice, supporting the role of TGF-β1 in promoting PH through enhanced cellular proliferation.[115,116]
Several lines of evidence suggest that PPARγ activation can modulate TGF-β1 signaling through Smad-dependent and Smad-independent mechanisms. For example, both natural (15d-PGJ2) and synthetic (ciglitazone and rosiglitazone) PPARγ ligands inhibited the profibrotic effects of TGF-β1 on human lung fibroblasts in a Smad-independent manner as evidenced by blunted myofibroblast differentiation and collagen I synthesis.[117] Adenoviral PPARγ gene overexpression similarly decreased fibroblast to myofibroblast differentiation, collagen I and III production in alkali burned mouse corneas.[118] In this study, PPARγ overexpression also reduced TGF-β1mRNA transcription in fibroblasts, cultured macrophages, and epithelial cells. In other studies, PPARγ agonists prevented TGF-β1 induced mesangial and hepatic stellate cell activation and extracellular matrix secretion.[119,120] Finally, PPARγ activation inhibited TGF-β1-mediated Smad 3 phosphorylation and induction of connective tissue growth factor expression, a key regulator of extracellular matrix production and neointima formation following vascular injury.[121] These findings emphasize that PPARγ modulation of TGF-β1 signaling is cell type and context specific and that PPARγ agonists may blunt abnormal TGF-β1/BMP-2 signaling and resultant pulmonary vascular remodeling in PH. Additional pathways by which PPARγ regulates cellular proliferation and vascular remodeling are summarized in Figure 4.
Matrix alterations
PH involves remodeling of pulmonary vessels with muscularization of nonmuscular distal arterioles, proliferation and migration of VSMC, and increased production of extracellular matrix proteins including fibronectin, collagen, and elastin.[122,123] Alterations in matrix composition may be related to increased matrix degradation resulting from an imbalance in the matrix metalloproteinases-tissue inhibitor of metalloproteinases system (MMP-TIMP) with deposition of collagen, elastin, fibronectin, and tenascin-C.[122] Inhibition of MMPs and elastase prevented the progression and actually induced regression of vascular remodeling in an experimental model of PH.[122] In addition, the elastase inhibitor, elafin, protected mice from chronic hypoxia-induced PH.[124] Recent evidence indicates that the PPARγ ligand, rosiglitazone, attenuated and reversed vascular remodeling in rat and mouse models of chronic hypoxia-induced PH.[15,21] Rosiglitazone decreased collagen production and elastin deposition and increased MMP-2 activity. Treatment with PPARγ ligands has also been associated with attenuation of matrix deposition in disorders other than PH.[125–127] Taken together, these reports suggest that therapeutic mechanisms of PPARγ activation in PH could involve attenuation of matrix deposition and remodeling in the pulmonary vasculature.
Inflammation
Several lines of evidence implicate inflammation in the pathobiology of PH. Despite the heterogeneity of disease conditions that lead to PH, similar inflammatory cells types can be found in the plexiform vascular lesions in patients with IPAH and PH associated with connective tissue disease and HIV. Perivascular mononuclear cell infiltrates comprising macrophages, T and B lymphocytes, and dendritic cells are reported in patients with PH.[64,66,68] Circulating markers of inflammation are also increased in patients with PH.[128] Proinflammatory cytokines and growth factors such as IL-1, IL-6, PDGF, epidermal growth factor (EGF), and vascular endothelial growth factor (VEGF) are increased and contribute to mitogenic and chemoattractant events in the vascular wall in PH.[64] Chemokines that mediate inflammation by recruiting leukocytes, monocytes, and T cells to the vascular wall are increased in patients with PH. Chemokines, including fractalkine (CX3CL1), regulated upon activation, normal T-cell expressed and secreted (RANTES), and monocyte chemotactic protein-1 (MCP-1), also promote pulmonary vascular remodeling in PH.[64,68] Fracktalkine is directly associated with pulmonary artery SMC proliferation in MCT-induced PH in the rat,[129] and both RANTES[68] and MCP-1[130] may indirectly contribute to mitogenesis and vasoconstriction by inducing endothelin converting enzyme-1 (ECE-1) and ET-1.
The stimulation of these inflammatory pathways and their modulation by PPARγ likely occurs at the level of transcriptional regulation. The transcription factor, NFAT, is upregulated in the pulmonary artery walls and circulating inflammatory cells of PH patients. NFAT, a master activator of T cells, regulates the expression of many inflammatory genes. In PAH, increases in NFAT confer a proliferative and apoptosis resistant phenotype on PASMC by increasing bcl-2, mitochondrial hyperpolarization, and downregulating voltage-gated potassium channels (Kv1.5).[131] Common stimuli of PH such as hypoxia cause inflammatory responses in the pulmonary vasculature. PPARγ is expressed in T cells, macrophages, leukocytes, and dendritic cells, suggesting that its activation could modulate inflammation in the pulmonary vasculature that contributes to the development of PH. Studies demonstrate that PPARγ ligands attenuate inflammation in numerous models.[132] PPARγ activation reduced macrophage recruitment and inflammatory mediator production, impaired dendritic cell priming of T cells, as well as T lymphocyte proliferation and viability. PPARγ ligands also induced regulatory T cells which downregulate immune responses.[133,134] Current evidence indicates that these anti-inflammatory effects of PPARγ are mediated not through transactivation of specific target genes, rather through physical binding to other proinflammatory transcription factors such as NF-kB, AP-1, and STAT, leading to suppression of these proinflammatory transcriptional pathways. The precise mechanisms of these transrepression effects remain to be completely defined but may involve ligand-dependent SUMOylation of PPARγ which targets the receptor to corepressor complexes on the promoters of inflammatory genes preventing proteosomal degradation of the corepressors and thereby causing inhibition of inflammatory gene expression.[10]
Apoptosis
Apoptosis is an important process whereby selected cells are programmed for death and removal.[135] Apoptosis counterbalances cell proliferation and mitotic division in a cascade of events involving caspase activation and protein cleavage, nuclear DNA fragmentation, cytoplasmic condensation, cell fragment sequestration and formation of apoptotic bodies.[136] Ineffective apoptosis contributes to the pathology of diseases including some malignancies, autoimmunity and persistent infections. Deregulated apoptosis, on the other hand, may be implicated in atherosclerosis and other cardiovascular diseases.[136] Triggers for apoptosis include, but are not limited to Fas death receptor stimulation by Fas ligand, recognition of cells with DNA damage, abnormal cellular migration and proliferation, ROS, angiotensin type 2, and BMP-2.[135]
The pulmonary artery SMCs of patients with PAH demonstrated increased tendency to proliferate and exhibited resistance to normal apoptotic signals such as bone morphogenetic protein-2 (BMP-2).[137] Accordingly, germline mutations in BMPR-2 significantly contribute to disease pathogenesis in 70% of patients with familial PAH.[109] Impaired apoptosis and proliferation of PASMC may result from increases in the apoptosis inhibitor, survivin, and the sequential occurrences: hyperpolarization of mitochondria, increased HIF-1α transcription and nuclear translocation, and decreased Kv1.5 voltage-gated potassium channel expression.[131] These derangements in the regulation of apoptosis in pulmonary vascular wall cells likely contribute to the increased cell proliferation and migration characteristic of the remodeled pulmonary vasculature in PH.[135] PPARγ agonists promote apoptosis of proliferating VSMCs in an extracellular signal-related kinase 1/2 (ERK 1/2) – independent manner that may involve interferon regulatory factor-1 (IRF-1) or activation of the proapoptotic genes, p53 and Gadd 45.[138,139] PPARγ agonists also induce apoptosis in endothelial cells in a PPARγ-dependent manner.[140] Similarly, pulmonary artery endothelial cells derived from patients with IPAH displayed a proliferative, apoptosis-resistant phenotype associated with enhanced STAT 3 signaling, and PPAR ligands reduced STAT 3 signaling.[141,142] Taken together, these studies indicate that multiple pathways can lead to an apoptosis-resistant phenotype in pulmonary vascular wall cells during the development of PH and that PPARγ may counterbalance these proliferative pathways by stimulating apoptosis.
Progenitor cell recruitment
Endothelial progenitor cells (EPCs) are bone marrow-derived cells that are mobilized into the systemic circulation in response to ischemia or vascular injury.[143] The role of EPCs in vascular repair, vascular homeostasis, and formation of new blood vessels continues to be defined.[144] Clinical trials suggest that EPCs may serve as markers of vascular dysfunction and cardiovascular disease.[145,146] Because endothelial dysfunction participates in the pathogenesis of PH, EPCs may play a role in pulmonary vascular disease.[147] The endogenous erythropoietin system recruits EPCs to the lung in experimental PH in mice.[148] Furthermore, mesenchymal stem cells overexpressing eNOS or EPCs expressing adrenomedullin attenuated MCT-induced PH in rats.[149] These reports suggest that migration of EPCs to the pulmonary vasculature during experimental PH can exert beneficial effects. PPARγ agonists facilitate the differentiation of angiogenic progenitor cells into EPC.[150] PPARγ agonists also increased the number and migratory activity of EPC in patients with type 2 diabetes and impaired endothelial function and increased EPC migratory activity and reduced EPC apoptosis in mice.[151] Collectively, these reports suggest that PPARγ ligands might stimulate EPC to reduce pulmonary vascular dysfunction and reduce PH.
Thrombosis in PH
Pulmonary artery thrombosis is a common pathologic finding occurring in 48-56% of patients with IPAH.[152,153] Thrombosis can also be found in other forms of PH associated with collagen vascular disease, HIV, portal hypertension, drugs and anorexigens. Thrombotic lesions in IPAH can be eccentric or concentric and usually occur in situ in peripheral muscular arteries where they form lesions that result from mural organization of thrombi.[49 153] Thrombosis is in large part due to abnormal platelet activation and endothelial dysfunction which results in coagulation and loss of counterbalancing antifibrinolytic mechanisms.[152,153] Shear stress from elevated pulmonary pressures may contribute to endothelial injury and release of mediators of coagulation.[49] Accordingly, patients with IPAH have increased serum levels of coagulation mediators including plasminogen activator inhibitor type-1 (PAI-1),[154] Von Willebrand factor (vWF),[155] fibrinopeptide A,[156] and factor VIIIc.[157] The abnormal platelet activation in PH can be attributed to increases in platelet-derived thromboxane (TXA2) and decreased levels of endothelial derived prostacyclin (PGI2) and nitric oxide synthase (eNOS).[158] Collectively, these imbalances favor coagulation and platelet aggregation which ultimately predisposes patients with PH to the development of thrombosis.
PPARγ agonists modulate platelets' immunoregulatory and proinflammatory functions and favorably affect vascular patency. Platelets release a variety of proinflammatory mediators and cytokines including prostaglandin E1, TGF-β, IL-1B, PAI-1, and CD 40.[48] For example, the CD 40 ligand is a transmembrane protein expressed on stimulated CD4+ T cells and platelets,[159,160] whose expression can be reduced by PPARγ ligands.[48,161] CD 40 ligand is associated with platelet activation and increased risk for cardiovascular disease.[48,159,160] Upon platelet activation, the CD 40 ligand is released and promotes the expression of vasoactive, inflammatory, and thrombotic mediators including cyclooxygenase-2 (COX-2), prostaglandins, TNF-α, IFN-γ, tissue factor, MMPs, selected interleukins, chemokines, and multiple adhesion molecules.[48,159–161] Thus, CD 40 receptor-ligand binding links platelet activation and vascular inflammation to hemostasis and increased risk for intravascular thrombosis. In vitro, thiazolidinediones decreased platelet CD 40 ligand expression and release.[159] In vivo experiments confirmed that diabetic mice treated with pioglitazone had decreased CD 40 expression after platelet activation.[162] These studies demonstrate that PPARγ agonists modulate platelet immunoregulatory and hemostatic function by attenuating CD 40 expression.
Since PPARγ belongs to a nuclear receptor superfamily of transcription factors, historically PPARγ expression was thought to be limited to nucleated cells. However, Akbiyik and colleagues reported that platelets contain PPARγ.[159] Given that platelets have no nucleus and express no PPARγ mRNA, PPARγ agonists may affect platelet function through non-transcriptional pathways such as modulation of intracellular signaling or platelet–protein interactions. For example, two studies reported that pioglitazone delayed the onset of iatrogenic arterial thrombosis in animal models.[163,164] The mechanisms for these PPARγ effects may include attenuation of platelet activation as evidenced by reductions in soluble and platelet bound P-selectin,[48,162] CD 40 ligand,[49,159] and TXA2,[49,159] or to direct effects of PPARγ ligands on vascular endothelium and its production of platelet regulators. PPARγ agonists also decreased adenosine diphosphate and arachidonic acid-induced platelet aggregation,[159,163] ATP production,[163] and increased nitric oxide-mediated activation of fibrinolysis and inhibition of coagulation.[163,165] Other proposed anti-thrombotic mechanisms involve PPARγ-induced increases in prostacyclin,[164] thrombomodulin,[163] as well as decreases in PAI-1 and fibrinogen.[166,167] In summary, PPARγ ligands likely decrease platelet activation and aggregation through direct effects on platelets and cells of the vascular wall that stimulate endothelial and platelet-derived vasodilatory mediators to maintain vascular patency and blood flow.
CONCLUSIONS AND FUTURE DIRECTIONS
The evidence reviewed above suggests that PPARγ can participate in the regulation of numerous pathways implicated in PH pathogenesis. Activation of PPARγ can reduce vasoconstriction, vascular remodeling and inflammation, and thrombosis that contribute to the generation and progression of PH. Studies employing animal models of PH have demonstrated that tissue-targeted deletion of PPARγ in vascular wall cells can promote PH while activation of PPARγ with exogenous ligands attenuated PH or vascular remodeling in MCT- or hypoxia-induced rodent models of PH.[14,15,17,20–22] The limitations of these animal models have been recently reviewed emphasizing that the performance of pharmacological tools in the treatment of common experimental models of PH in rodents may not accurately translate to the treatment of human PAH.[168] The investigation of PPARγ ligands in recently reported rodent models which more closely reproduce pathological changes seen in the pulmonary arteries of patients with advanced PAH provides an experimental strategy that might circumvent some of the limitations of common existing models.[169]
Based on the findings in this review, the next step in the evaluation of PPARγ as a therapeutic target in PH would appear to be clinical trials employing PPARγ ligands in patients with PH. The abundant evidence reviewed above that PPARγ ligands can favorably modulate many pathological pathways and mediators involved in PH would support this position. In addition, the current availability of synthetic thiazolidinedione PPARγ ligands (rosiglitazone and pioglitazone) for the treatment of type 2 diabetes in the United States suggests that clinical trials could be expedited. However, recent findings surrounding potential adverse cardiovascular events in diabetic patients taking rosiglitazone emphasize the need for caution before employing this agent in PH patients with preexisting cardiopulmonary disease.[170,171] In contrast, clinical studies with pioglitazone in diabetic patients have reported a lowered risk for adverse cardiovascular endpoints.[172–175] These reports emphasize that individual PPARγ ligands may regulate unique patterns of gene expression that differentially modulate cell and tissue function. Thus, the therapeutic potential of strategies targeting PPARγ will be optimized by future studies that must determine not only the relevant molecular pathways that are altered by PPARγ in a ligand-specific manner, but also the cellular site of action of any ligand and its relative dependence on the PPARγ receptor. Evidence supporting successful therapy of PH with existing PPARγ ligands could also stimulate the development of novel pharmacological PPARγ ligands with enhanced therapeutic efficacy and/ or reduced side effects.
Footnotes
ACKNOWLEDGMENTS
The authors acknowledge grant support from the Research Service of the Atlanta Veterans Affairs Medical Center and the National Institutes of Health (R01 DK 074518). Dr. Green is supported by an NHLBI T32 training grant (HL076118-06).