
Abstract
Mutations in the bone morphogenetic protein type II receptor gene (BMPR-II) are the major cause of heritable pulmonary arterial hypertension (PAH). Although both endothelial and smooth muscle cell BMPR-II dysfunction have been seen to contribute to pulmonary hypertension in vivo, little is known about the impact of BMPR-II mutation on the interaction between these two important cell types. We employed adenoviral vectors to overexpress wild type or mutant (kinase-deficient mutation, D485G) BMPR-II in human pulmonary arterial endothelial cells (PAECs). PAECs transfected with mutant BMPR-II demonstrated increased susceptibility to apoptosis. Conditioned media from PAECs transfected with mutant BMPR-II increased the proliferation of pulmonary arterial smooth muscle cells (PASMCs), when compared with conditioned media from PAECs transfected with wild-type BMPR-II. PAECs transfected with mutant BMPR-II released higher levels of TGF-β1 and FGF2 into the conditioned media than the wild-type BMPR-II-transfected cells. Conditioned media from PAECs expressing mutant BMPR-II also showed increased activation of luciferase activity in a TGF-β bioassay. The increased proliferation observed in PASMCs exposed to conditioned media from PAECs expressing mutant BMPR-II was inhibited by neutralizing the antibodies to TGF-β1, or small molecule inhibitors of ALK-5 (SD208) or FGFR1 (SU5402). We conclude that mutation in BMPR-II increases susceptibility to apoptosis of PAECs and leads to secretion of growth factors that stimulate the proliferation of PASMCs. These processes may contribute to the remodeling of pulmonary arteries observed in patients with familial or heritable PAH.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by a marked increase in pulmonary arterial pressure and right ventricular hypertrophy Without treatment, progression of pulmonary hypertension leads to right ventricular failure and death in ~three years from diagnosis.[1] Mutations in the gene, encoding the bone morphogenetic protein type II receptor (BMPR-II),[2,3] have been found in ~70% of families with heritable or familial PAH;[4]and up to 25% of the patients with apparently sporadic or idiopathic PAH (IPAH) harbor mutation.[5] BMPR-II is highly expressed on the vascular endothelium of the pulmonary arteries and at a low level in PASMCs and fibroblasts in the lung.[6]
Endothelial cells (ECs) are recognized as major regulators of vascular function, balancing the production of vasoconstrictors versus vasodilators, activators versus inhibitors of smooth muscle cell (SMC) growth and migration, prothrombotic versus antithrombotic mediators, and proinflammatory versus anti-inflammatory signals.[7,8] Recent studies have shown that apoptosis of ECs induces the release of mediators that cause vascular SMC proliferation. [9] Endothelial dysfunction in PAH may allow excessive release of paracrine factors that act either as growth factors to induce PASMC proliferation or as chemokines to recruit circulating inflammatory cells.[10–12] Exposure of pulmonary arterial SMCs (PASMCs) to the culture medium from pulmonary arterial ECs (PAECs) induces PASMC proliferation, and this effect is exaggerated when PAECs from patients with PAH are used.[10] However, the role of endothelial dysfunction caused by the mutation of BMPR-II on PASMC hyperplasia remains unclear.
In the present study, we examined the impact of introducing a kinase-inactive, disease-relevant BMPR-II mutant into the PAECs and demonstrated that this promotes apoptosis of the PAECs. This was associated with an increased release of TGF-β1 and FGF2 from the PAECs transfected with mutant BMPR-II. Furthermore, the conditioned media from mutant BMPR-II-transfected PAECs stimulated a heightened proliferation of PASMCs, an effect that could be inhibited with a neutralizing antibody to TGF-β1 or an ALK-5 inhibitor, or a small molecule inhibitor of the fibroblast growth factor receptor-1 (FGFR1). These findings demonstrated that the mutation in BMPR-II promoted the apoptosis of PAECs and stimulated the release of growth factors, with pro-proliferative effects on PASMCs. These findings provided a mechanism by which the BMPR-II mutation drove the cellular processes that contributed to pulmonary vascular remodeling in patients with heritable PAH.
MATERIALS AND METHODS
Cell culture and experimental design
Human pulmonary artery endothelial cells (PAECs; n=3) and an EGM-2 Bullet Kit were purchased from Lonza (UK). PAECs were grown in 5% fetal bovine serum (FBS) / EGM-2 and used between passage 5~6.[13] The cells were plated at 2.5×105 cells in 60-mm culture dishes or 0.5×104 cells in eight-well chamber slides, for adenoviral transfection. The control and mutant human pulmonary artery smooth muscle cells (PASMCs, n=3 in each group) were explant-derived from the peripheral pulmonary arteries (<2 mm external diameter), as previously described (14). The Papworth Hospital (UK) Ethical Review Committee approved the study, and the subjects or their relatives gave an informed written consent. The cells were maintained in 10% FBS / Dulbecco's modified Eagle Medium (DMEM) and used between passages 4~6. The smooth muscle phenotype of isolated cells was confirmed, as previously described.[14]
Adenoviral transfection
The generation and transfection of the adenoviral constructs was described as previously expressed.[13] In brief, the PAECs were plated in 60-mm dishes or eight-well chamber slides in cultured medium, overnight. After washing in serum-free EGM-2 twice, the cells were transfected with overexpress wild-type (Wt) or mutant (kinase-deficient mutation, D485G) of BMPR-II constructs for 48 hours at 50 pfu / cell, in serum-free EGM-2.
Apoptosis assay
Apoptosis was assessed by a morphological assay after H-33342 and propidium iodide (PI) staining, as previously described.[14] Flow cytometric analysis was also used to determine apoptotic cells, using an FITC Annexin V staining kit (BD Biosciences, UK) according to the manufacturer's protocol. Briefly, after 48 hours of adenoviral infection, PAECs were washed thrice with serum-free EGM-2 (SFE2). The cells were maintained in serum-free EGM-2, after the last wash, for 24 hours. A serum-free EGM-2 without growth factors (SF) was used as the positive control for apoptosis. The cells were harvested by brief trysinization and washed twice with cold PBS. Five microliters of Annexin V-fluorescein isothiocyanate (FITC) and PI were added to every 100 μl of cell suspension, for 15 minutes, at room temperature, in the dark, and then 400 μl of 1x binding buffer were added to each sample before analysis. FACS analysis was carried out using a Becton Dickinson FACS Scan analyzer (BD Biosciences, UK).
Conditioned medium
After a 24-hour incubation in serum-free EGM-2, the medium in 60-mm dishes was collected from the PAECs transfected with adenoviral BMPR-II constructs (Wt and D485G), and designated as CMWt and CMMut, respectively. The medium from nontransfected PAECs incubating in serum-free EGM-2 alone for 24 hours was used as the control (CMControl). Conditioned media were centrifuged at 15,000 g for 5 minutes at 4°C. The supernatants were then aliquoted and stored at −80°C and thawed for use in subsequent experiments.
Proliferation of PASMCs in response to conditioned medium
Pulmonary arterial smooth muscle cell proliferation was quantified by cell number counting and by 3H-thymidine incorporation assay, as previously described.[14] In brief, PASMCs were plated at 1.5×104 cells / well in 24-well plates, overnight. The following day, the cultured medium (CM) was replaced with 0.5% FBS in CM, with or without pretreatment, with a neutralizing antibody to TGF-βa (NA-TGF, 10 μg/ml; R and D Systems, UK). Further experiments involved incubation with the ALK-5 receptor inhibitor, SD208, (0.5 μM; TOCRIS, UK), or SU5402 (10 μM; Calbiochem, UK), a small molecule inhibitor of FGFR1. The 3H-thymidine was added for the last six hours. The cell counts were carried out on day 2 or 3, and day 5. The conditioned medium was replenished on day 3. Each experiment was repeated at least thrice.
Enzyme-linked immunosorbent assay (ELISA)
Sandwich ELISA kits (R and Sytems, UK) were used to measure the amount of TGF-β1 or FGF2 in the conditioned medium following the manufacturer's protocol. The samples were read using a spectrophotometer at an absorbance of 405 nm.
Luciferase activity assay
Mink lung epithelial cells (MLECs-clone 320) were used to quantify the active TGF-β1 in a conditioned medium.[15] The MLECs (provided by Dr. D.B. Rifkin, Department of Cell Biology, New York University Medical Center) were stably transfected with human plasminogen activator inhibitor-1 promoter-luciferase construct (PAI-L). Mature TGF-β got bound to the receptors of the MLECs resulting in a dose-dependent increase in luciferase activity. Briefly, the cells were seeded at 1.5×104 / well in 96-well plates, in 10% FBS/DMEM with geneticin (0.25mg/ml), for three hours. Next the medium was replaced by 100 μl of the control or conditioned medium for 20 hours. The cells were lysed in 100 μl / well of lysis buffer. The luciferase activity was assayed according to the manufacture's instruction (Roche, UK).
Statistics
Data were expressed as mean±SEM and analyzed with GraphPad Prism version 5.01 (GraphPad Software). Comparisons were made by Students t-test or ANOVA, as appropriate, and a value of P<0.05 indicated the statistical significance.
RESULTS
Overexpression of mutant BMPR-II causes apoptosis of PAECs
We used two methods (nuclear morphology and flow cytometry) to investigate the effects of overexpressing mutant BMPR-II (D485G) on apoptosis of PAECs. First we compared the PAEC number in non-transfected cells and in PAECs transfected with wild-type (Wt) or D485G BMPR-II (Mut) for 48 hours. There was a trend toward reduced cell number in cells transfected with Mut [Figure 1a and b]. The nuclear morphology and quantification demonstrated that after incubation in serum-free EGM-2 (SFE2) for 24 hours, the PAECs transfected with mutant BMPR-II exhibited a higher nuclear condensed cell number (27.9±5.7%) compared with Wt (7.4±1.9%) and control cells (4.8±2.1%) [Figure 1c and d]. The increased apoptosis was further demonstrated by flow cytometric analysis. The percentage of apoptotic cells (early and late-stage apoptosis) was increased following the incubation of cells in SFE2 without growth factors (SF) [Figure 2a and b]. The transfection of cells with the Wt construct did not change the percentage of apoptotic cells compared with SFE2 (12.6±3.2%) [Figure 2c]. In contrast, transfection of PAECs with the D485G construct increased the percentage of apoptotic PAECs significantly (25.7±7.1%) [Figure 2d]. The differences are shown graphically in [Figure 2e].
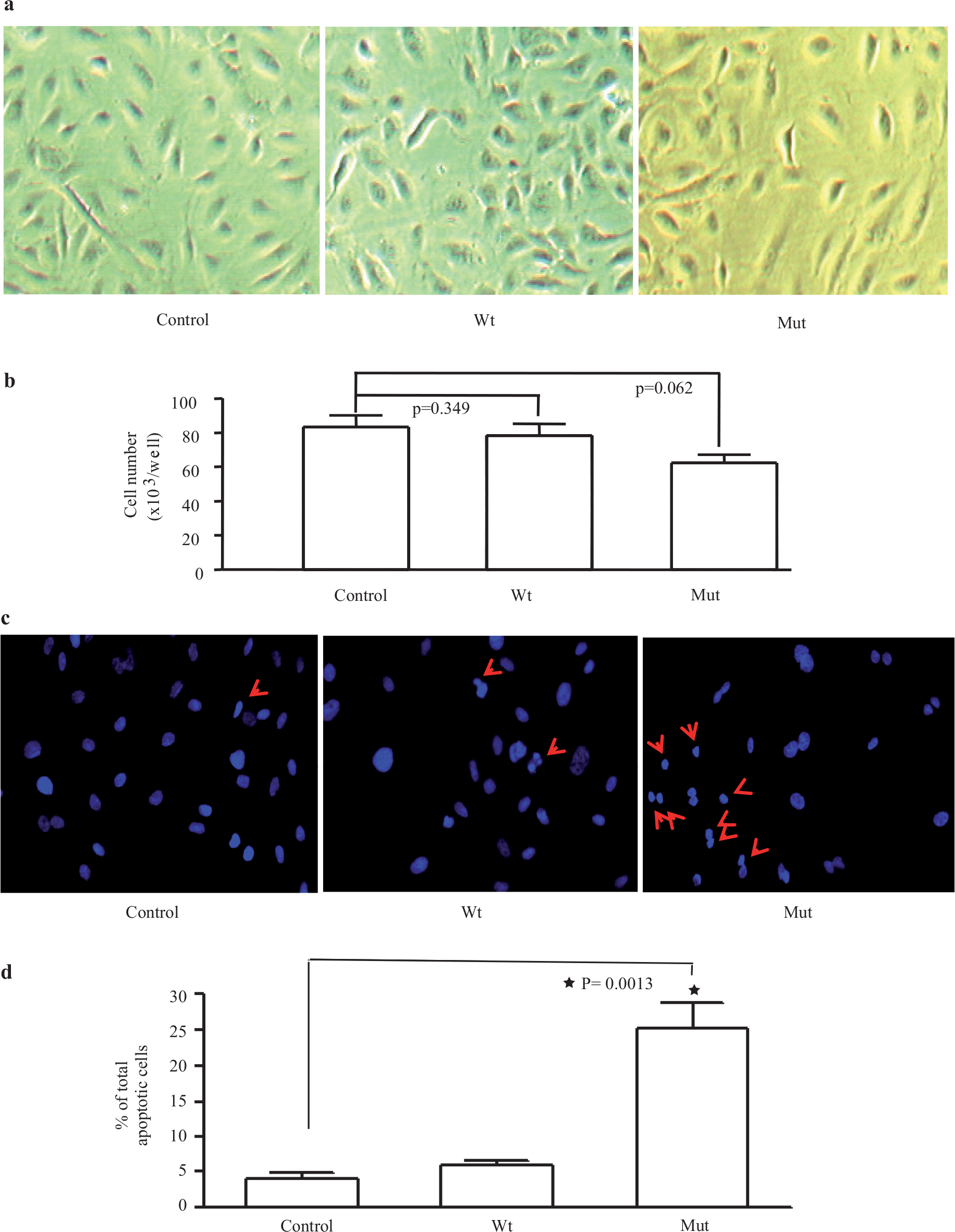
Pulmonary artery endothelial cell morphology and number, 48 hours following adenoviral transfection with wild type (Wt) or D485G (Mut) BMPR-II constructs, or non-transfected (Control) conditions (a and b). Following H-33342 staining for nuclear morphology (c) (red arrows indicates apoptotic cells) quantification demonstrated that after incubation in serum-free EGM-2 for 24 hours, the PAECs transfected with mutant BMPR II exhibited a higher nuclear condensed cell number (27.9±5.7%) compared to wild-type cells (7.4±1.9%) and NS (4.8±2.1%) (d)
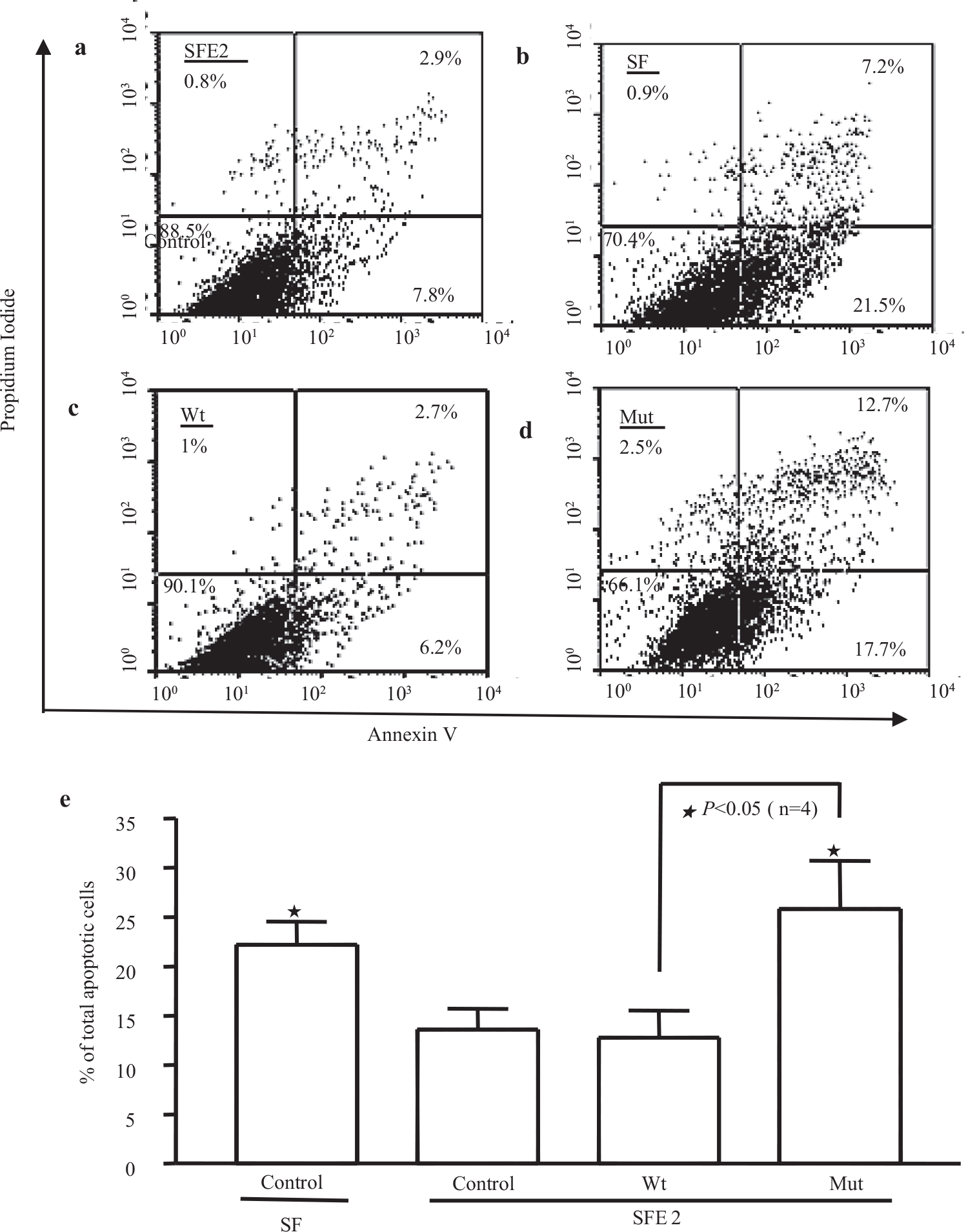
Apoptosis was further measured by flow cytometric analysis. (a, b, c, and d) Represents scatter plots of propidium iodide (PI) (y-axis) versus Annexin V-FITC (x-axis). Lower left quadrants (absence of both markers) indicate viable cells; upper left quadrants (PI positive) indicate cellular necrosis. The total apoptotic cells (early and late-stage apoptosis) are represented by the right side of the panel (Annexin V staining alone or together with PI) in which the total cell death number from the Mut is significantly higher (25.7±7.1%) than the Wt (12.6±3.2%) (e)
Conditioned media from PAECs transfected with mutant BMPR-II stimulate proliferation of PASMCs
We employed 3H-thymidine uptake and cell counting to examine the growth rates of PASMCs (n=3) exposed to a conditioned medium. PASMCs incubated with a conditioned medium from PAECs transfected with Wt BMPR-II (CMWt) showed similar 3H-thymidine incorporation and growth rates compared to PASMCs exposed to conditioned media from non-transfected PAECs (CM control) [Figure 3a and b]. In contrast PASMCs exposed to conditioned media from PAECs transfected with mutant BMPR-II (CMMut) exhibited increased 3H-thymidine incorporation and increased rates of proliferation [Figure 3a and b].
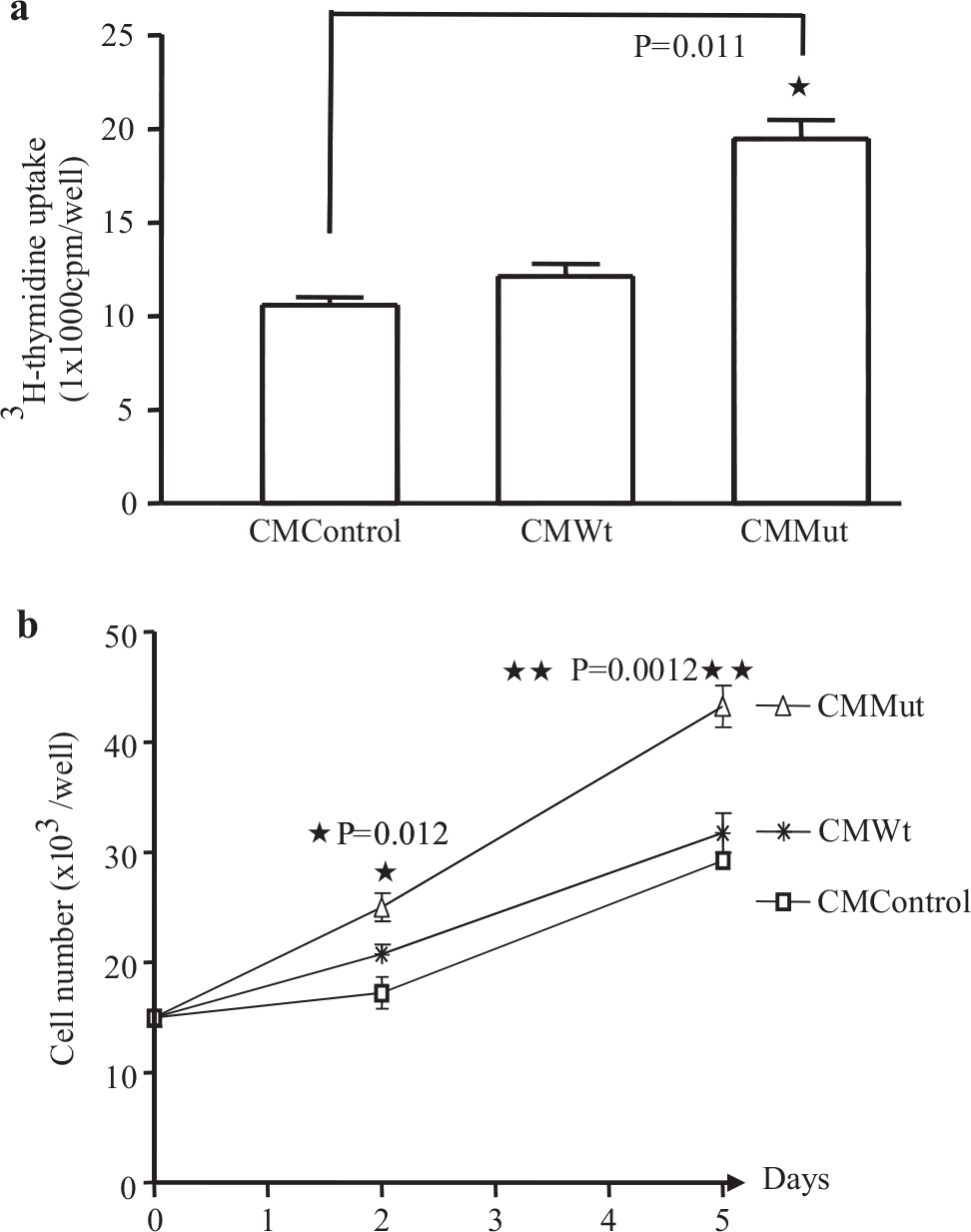
3H-thymidine uptake (a) and cell number (b) of PASMCs (n=3) in conditioned medium (CM) from PAECs. The PASMCs seeded in 0.5% FBS / CMMut for 24 hours show a significant increase in the DNA synthesis compared to the CM from control cells or CM from Wt transfected cells (a). Cell numbers were also increased after two days of incubation in 0.5% FBS/CMMut, a difference that was maintained at day 5 (b)
PAECs expressing mutant BMPR-II release higher levels of TGF-β1 and FGF2
Increased endothelial release of FGF2 was reported in PAECs from idiopathic PAH patients [12] and increased release of TGF-β1 was observed in endothelial cells undergoing apoptosis.[9] Both of these growth factors were known to play important roles in pulmonary vascular remodeling. We questioned whether conditioned media from PAECs transfected with mutant BMPR-II contained increased levels of these mediators. Employing ELISAs to detect the amount of TGF-β1 and FGF2 in the three sets of conditioned media, we observed higher levels of TGF-β1 and FGF2 in the media conditioned by PAECs transfectd with mutant BMPR-II, compared to WT transfected cells and non-transfected cells [Figure 4a and b]. In addition, we measured the TGF-β1 activity of the conditioned media using the MLEC bioassay. This assay further confirmed the finding of increased TGF-β1 activity in the media conditioned by PAECs transfected with mutant BMPR-II [Figure 4c].
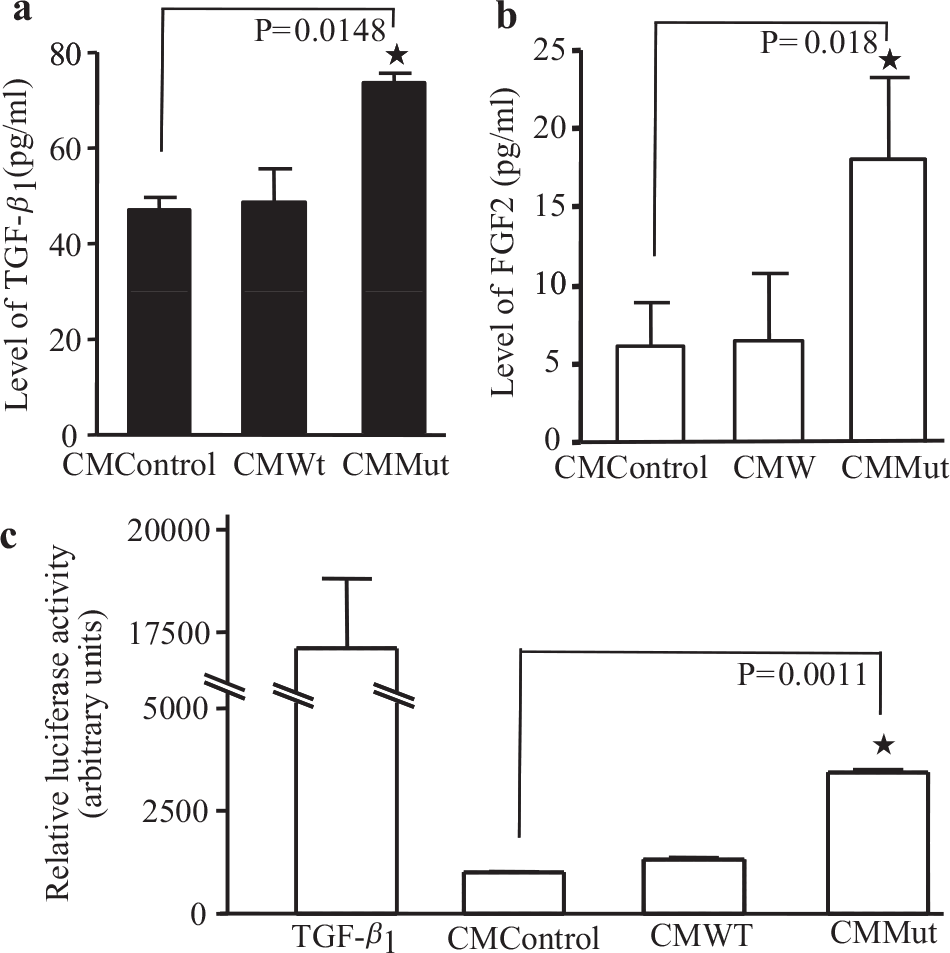
The level of TGF-β1 or FGF2 in the three types of conditioned medium were measured by ELISA. Both growth factors were higher in the conditioned media (CM) from the Mut cells compared to the CM from the control, or Wt transfected cells (a, b). Luciferase assays in the mink lung epithelial cells, stably expressing the human PAI-1 promoter, confirmed that CM from mutant transfected PAECs (CM Mut) contained more active TGF-β than CM from untransfected (control) cells or CM from PAECs transfected with WT BMPR-II (CM WT) (c). TGF-β 1 was used as a positive control in this assay
Pro-proliferative effects of mutant PAEC conditioned media are inhibited by inhibitors of ALK-5 and FGFR1
In order to confirm the functional significance of the observed increased levels of TGF-β1 and FGF2 in conditioned media from PAECs transfected with mutant BMPR-II, we performed further experiments employing inhibitors of TGF-β and FGF2 signaling. Thus pretreatment of control PASMCs with an anti-TGF-βx neutralizing antibody resulted in the inhibition of PASMC proliferation to PAEC mutant conditioned media [Figure 5a]. Similar effects were observed with an inhibitor of the TGF-β type 1 receptor (ALK-5), SD208 [Figure 5b]. A small molecule inhibitor of the FGFR1 receptor, SU5402, also markedly inhibited the pro-proliferative effect of conditioned media from PAECs transfected with mutant BMPR-II [Figure 5b]. The results of experiments using 3H-thymidine incorporation were mirrored by experiments employing cell counting [Figure 6a]. In the latter studies we also determined the effect of conditioned media on PASMCs isolated from PAH patients known to harbor mutations in BMPR-II (N903S; W9X; R899X). Although there was a trend to higher rates of proliferation in mutant PASMCs (~25%) compared with PASMCs from the control subjects, this was not statistically significant (P=0.062] [Figure 6a and b]. Conditioned media from PAECs transfected with mutant BMPR-II had a similar effect to that observed in control PASMCs, as did the inhibitors of ALK-5 and FGFR1
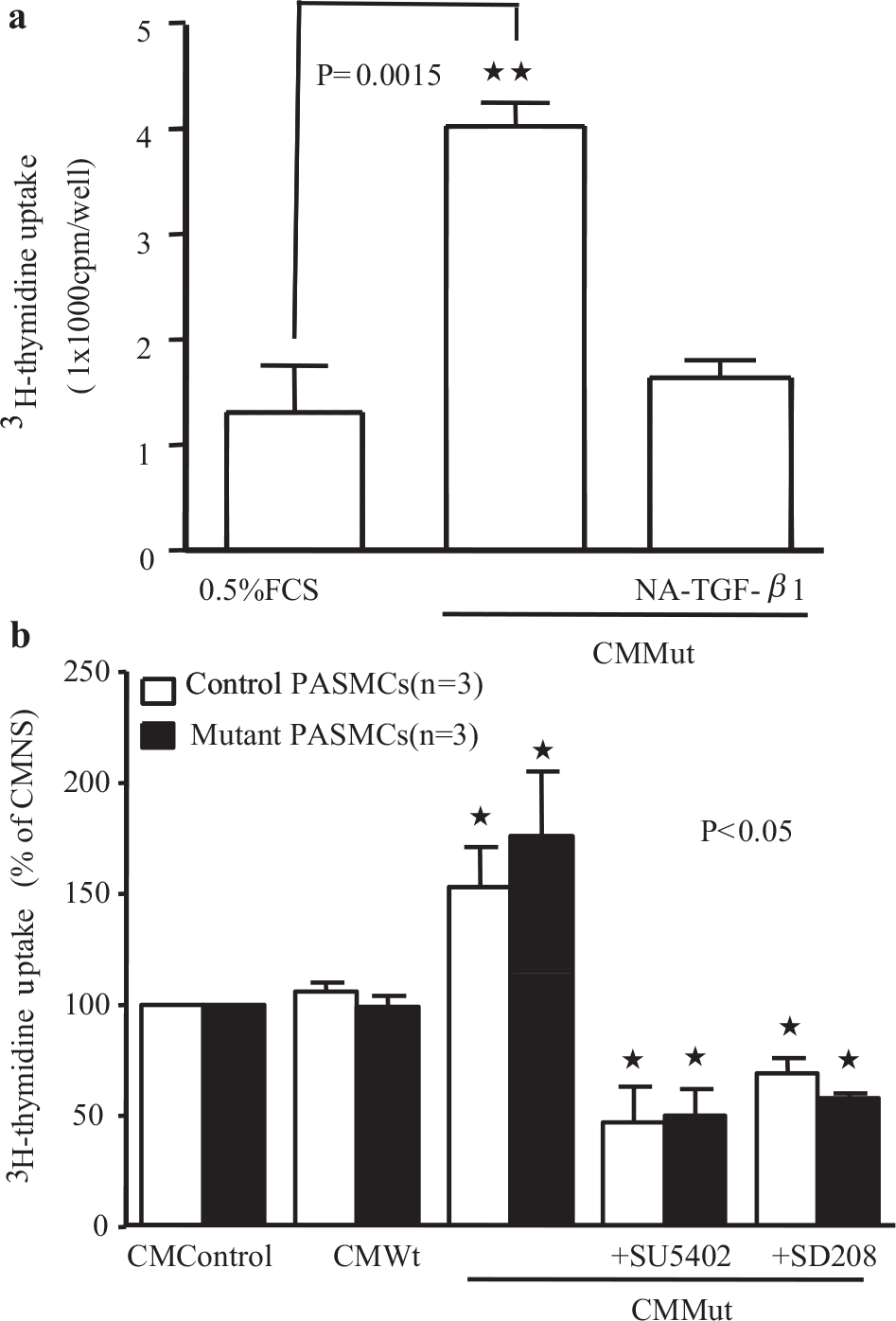
Graphs showing 3H-thymidine uptake in normal PASMCs following treatment with conditioned media (CM) from control PAECS, or PAECs transfected with WT or Mut BMPR-II constructs. A specific TGF-β1 neutralizing antibody significantly reversed the CM Mut-induced DNA synthesis in PASMCs (a). Similar effects were observed with an inhibitor of the TGF-β type 1 receptor (ALK-5), SD208, and the FGFR1 inhibitor, SU5402 (b)
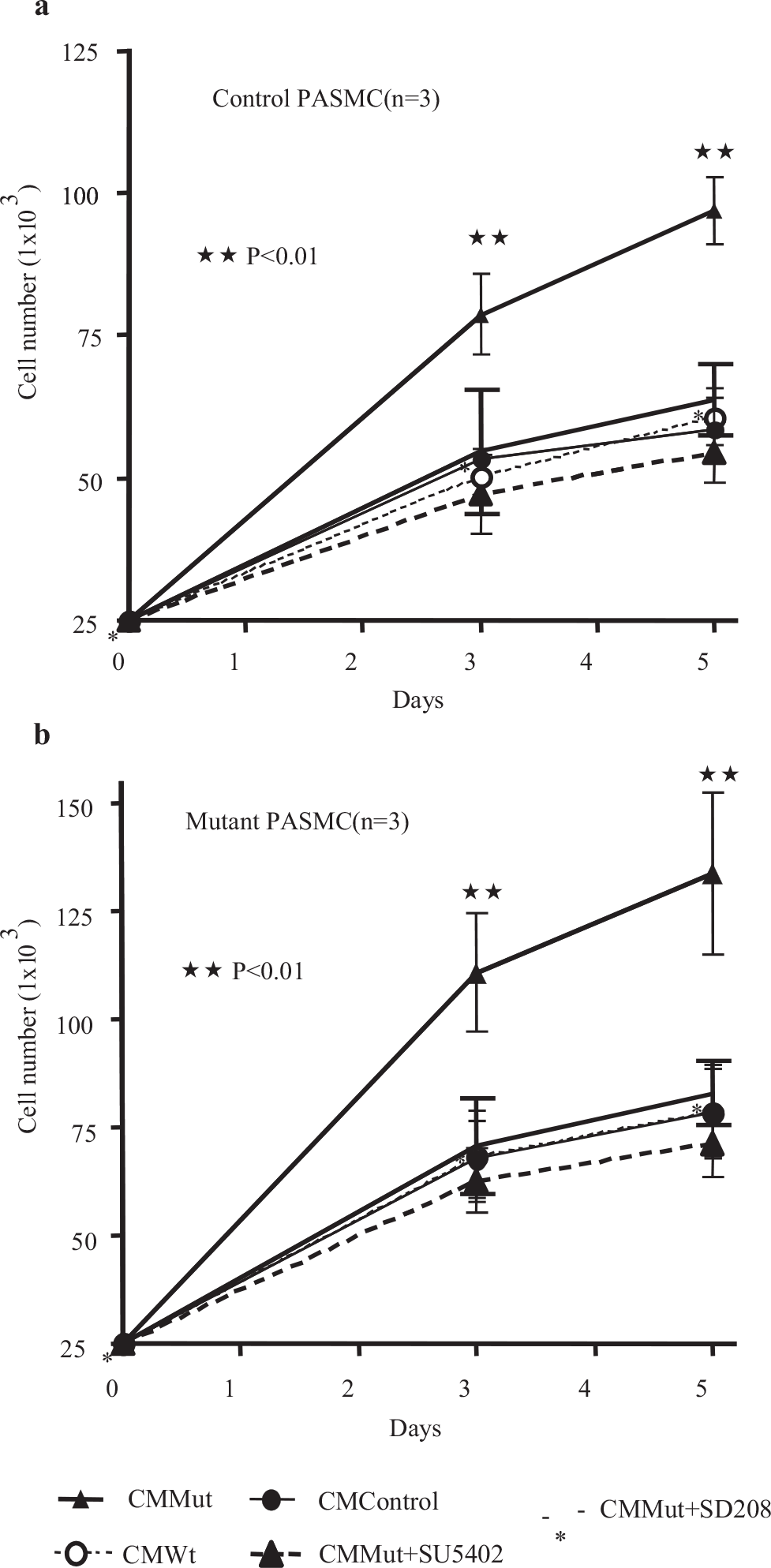
Graphs showing control (a) and BMPR II mutant (b) PASMC numbers following treatment with conditioned media (CM) from control PAECS, or PAECs transfected with WT or Mut BMPR-II constructs. The mutant PASMCs harbored one of the following mutations: N903S; W9X; or R899X. Increased proliferation was observed in both control and mutant PASMCs in the presence of CM from PAECs transfected with mutant (Mut) BMPR-II. There was a trend toward higher rates of proliferation (Figure 6b) in mutant PASMCs (~25%) compared to the control PASMCs, but this did not reach statistical significance (P=0.062). The results in the control and mutant PASMCs were otherwise similar and the heightened growth response to CM Mut could be abrogated by SU5402 or SD208 in both cell types
DISCUSSION
In this study we provide evidence that overexpression of mutant BMPR-II confers increased susceptibility to apoptosis in PAECs. These data support the observation that siRNA knockdown of BMPR-II also promotes endothelial apoptosis.[16] Endothelial apoptosis is considered to be a key trigger to disease initiation in the pathobiology of pulmonary hypertension.[7] The subsequent ingress of serum factors into the vessel wall leads to the activation of growth factors and of endogenous vascular elastases.[17] Here we report that an additional factor contributing to vascular remodeling may be the release of pro-proliferative growth factors from PAECs undergoing apoptosis.
Apoptotic cells are known to secrete TGF-β and this has also been shown for PAECs.[9] A previous report has also found that the released TGF-β from PAECs could stimulate proliferation of PASMCs.[9] In the present study we provide a link between BMPR-II mutation, endothelial apoptosis, and the secretion of growth factors from PAECS that can contribute to the proliferation of the underlying mesenchymal cells. In our study, overexpression of mutant BMPR-II in PAECs led to the release of both TGF-β1 and FGF2 and increased PAEC apoptosis. Increased endothelial apoptosis has also been noted in the lungs of heterozygous BMPR-II null mice exposed to inflammatory stimuli.[18]
Serum-free media conditioned by PAECs transfected with mutant BMPR-II and subjected to co-culture with control PASMCs, caused an abnormal proliferation of PASMCs. This pro-proliferative effect could be inhibited by a neutralizing antibody to TGF-β1 or an ALK-5 inhibitor (SD208), or a small molecule inhibitor (SU5402) of fibroblast growth factor receptor-1 (FGFR1). These findings provide a mechanism by which the BMPR-II mutation drives the cellular processes that contribute to pulmonary vascular remodeling in patients with heritable PAH.
Recent studies have demonstrated that BMP signaling represents an inhibitory pathway, which prevents excessive pulmonary arterial muscularization by reducing PASMC growth and increasing apoptosis.[14,19–21] In contrast, BMPs in general promote proliferation and are anti-apoptotic in endothelial cells.[13,16,22] Knockdown of BMPR-II with siRNA increases the susceptibility of PAECs to apoptosis.[16] The contrasting effects of BMPs in pulmonary vascular ECs and the underlying PASMCs provide a hypothesis for pulmonary vascular damage and remodeling in heritable PAH. A critical reduction in BMPR-II function in the endothelium may promote increased endothelial apoptosis, which compromises the integrity of the endothelial barrier and contributes to endothelial dysfunction. This view is almost certainly an oversimplification. In particular, BMP9 was recently identified as a ligand for a receptor complex, comprising of ALK-1 and BMPR-II.[23] In contrast to other BMPs, BMP9 appears to inhibit the proliferation of PAECs.[24] Therefore, the importance of diverse BMP ligands to the endothelial response in vivo remains to be elucidated.
Apoptosis and engulfment of apoptotic cells is known to be accompanied by the robust release of TGF-β.[25] Our group has previously shown that PASMCs isolated from patients with idiopathic PAH or heritable PAH exhibit an exaggerated growth response to TGF-β1 compared to control cells.[20] We therefore questioned whether the growth of PASMCs harboring BMPR-II mutations would be more susceptible than control cells to the pro-proliferative effects of conditioned media from PAECs transfected with mutant BMPR-II. Conditioned media from mutant transfected PAECS caused an increased proliferation of both control and BMPR-II mutant PASMCs. Although there was a trend towards heightened proliferation of PASMCs from patients harboring BMPR-II mutations, this did not reach statistical significance.
Taken together, our findings further strengthen the view that BMPR-II mutation promotes PAEC apoptosis and provide evidence that part of the endothelial dysfunction includes an increased release of growth factors that support proliferation of the underlying mesenchymal cells. Our data further suggests that BMPR-II mutation in PAECs leads to altered cross-talk between endothelial and smooth muscle cells that could contribute to the pathobiology of pulmonary hypertension.
Footnotes
ACKNOWLEDGMENTS
This project was funded by the British Heart Foundation (Program grant RG256 to NWM) and the European Commission, under the Sixth Framework Program (Contract No LSHM-CT-2005-018725, PULMOTENSION).