
Abstract
Although mild increase in pulmonary vascular resistance following intracardiac repair of tetralogy of Fallot is often seen in the early postoperative period, it usually subsides without any sequel. Persistent severe pulmonary artery hypertension after total correction is rare. We report a child with tetralogy of Fallot and Down's syndrome, who developed severe pulmonary hypertension and low cardiac output syndrome following an intracardiac repair, which was resistant to specific pulmonary vasodilators and increasing ionotropes. The post correction echocardiogram suggested an intact ventricular septal defect patch, no residual gradient across the right ventricular outflow tract, with free pulmonary regurgitation. The child had a poor outcome. A postmortem biopsy revealed histopathological signs of pulmonary hypertension.
INTRODUCTION
A mild increase in pulmonary vascular resistance is often seen after a successful total correction of tetralogy of Fallot (TOF) in the early postoperative period. This is usually reversed with time, without producing untoward consequences. Persistence of severe pulmonary artery hypertension (PAH) following corrective surgery for TOF is rare.[1] We present a case of TOF with Down's syndrome (DS), where there was development of severe PAH and low cardiac output syndrome following intracardiac repair (ICR), leading to a negative outcome. We also discuss the possible causes of PAH in this setting.
CASE REPORT
A seven-year-old male child, weighing 18 kg, presented to our Cardiac Surgical Outpatient Department, with a history of tachypnea and cyanosis since birth. His perinatal period was uneventful. General physical examination revealed features of DS with mental retardation and delayed developmental milestones, besides cyanosis and clubbing. The pulse rate was 82 per minute and blood pressure was 100 / 60 mmHg. On auscultation there was a 3 / 6 ejection systolic murmur in the pulmonary area, and normal breath sounds. Routine blood examination was normal except for hemoglobin of 15.3 gm%. Electrocardiogram revealed features of right ventricular hypertrophy. Chest X-ray showed pulmonary oligemia and a boot-shaped heart. The echocardiogram revealed a 15 mm perimembranous ventricular septal defect (VSD) with 50% aortic override, severe right ventricular outflow tract (RVOT) obstruction (peak gradient 110 mmHg), with infundibular, valvular, and supravalvular (small main pulmonary artery) stenosis. A small-sized (3 mm) patent ductus arteriosus (PDA), with a left-to-right shunt, and a left superior vena cava (LSVC) was draining into the coronary sinus. The right and left ventricular systolic function was normal. A preoperative catheterization study showed equal pressures in right ventricle (RV), left ventricle (LV), and aorta. The pulmonary artery (PA) was not entered. The coronary anatomy was normal and there were no aortopulmonary collaterals.
He underwent intracardiac repair with pericardial patch augmentation of the RVOT and main pulmonary artery, together with PDA ligation, and cardiopulmonary bypass (CPB). Total CPB time and aortic cross clamp time were 132 and 92 minutes, respectively. Termination of CPB was achieved using adrenaline 0.1 – 0.15 μg/kg/minute, dopamine 8 μg/kg/minute, and nitroglycerin 0.5 μg/kg/minute. Immediately after CPB, needle insertion in the RV and PA showed a pressure of 55 / 4 and 48 / 15 mmHg, respectively, with a corresponding systemic pressure of 65 / 44 mmHg. In the postoperative period, he developed features of low cardiac output syndrome with increase in ionotropic requirement. The postoperative ECG revealed atrioventricular block. Echocardiogram on day two showed dilated RA and RV, intact VSD patch, with no RVOT obstruction, and mild right pulmonary artery (RPA) origin stenosis. The catheterization study done on postoperative day two revealed the presence of severe PAH (RV systolic pressure – 55 / 8 mmHg, with a corresponding systemic pressure of 82 / 43 mmHg) with no gradient across the RVOT. Pulmonary artery angiogram showed mild RPA origin stenosis [Figure 1] and free pulmonary regurgitation. LSVC draining to the LA was ruled out [Figure 2]. He was started on sildenafil 0. 5 mg / kg every six hours and Thyroxin 12.5 μg once daily, besides an increase in inotropes. Furthermore, the postoperative course was characterized by hypotension in spite of maximal inotropes. Finally the patient had a cardiopulmonary arrest on postoperative day four and could not be revived. The subsequent postmortem lung biopsy revealed features of pulmonary hypertension, with medial hypertrophy and intimal proliferation in the intra-acinar arterioles corresponding to grade II of the Heath-Edwards grading system of pulmonary hypertension [Figure 3].
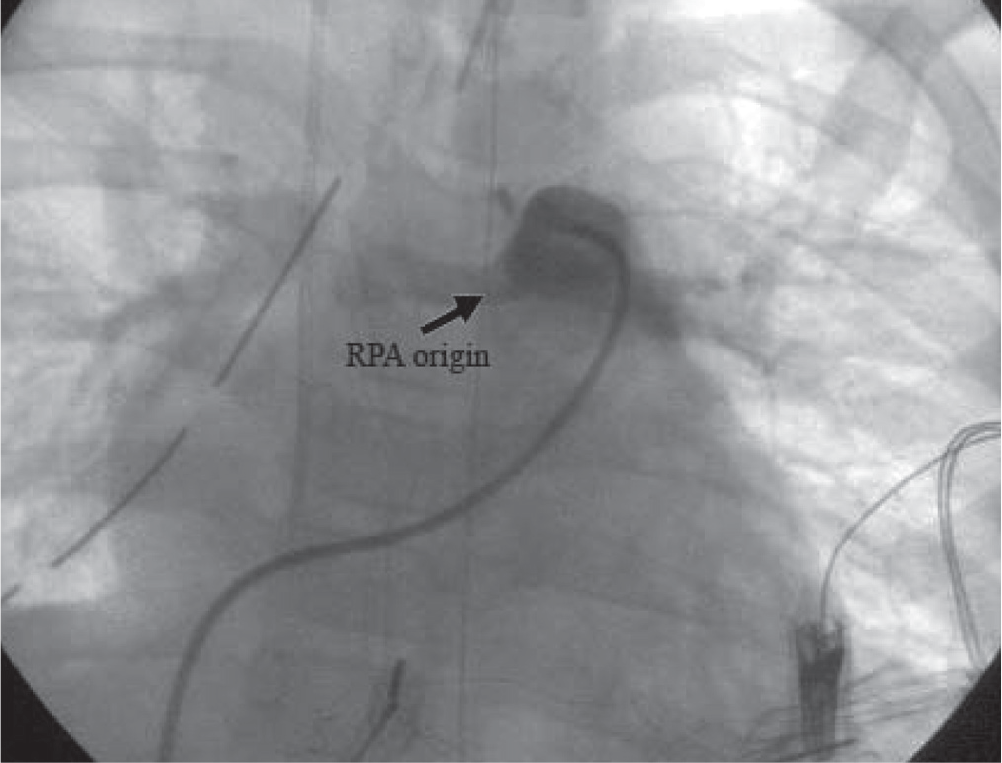
Catheterization study showing mild RPA origin stenosis
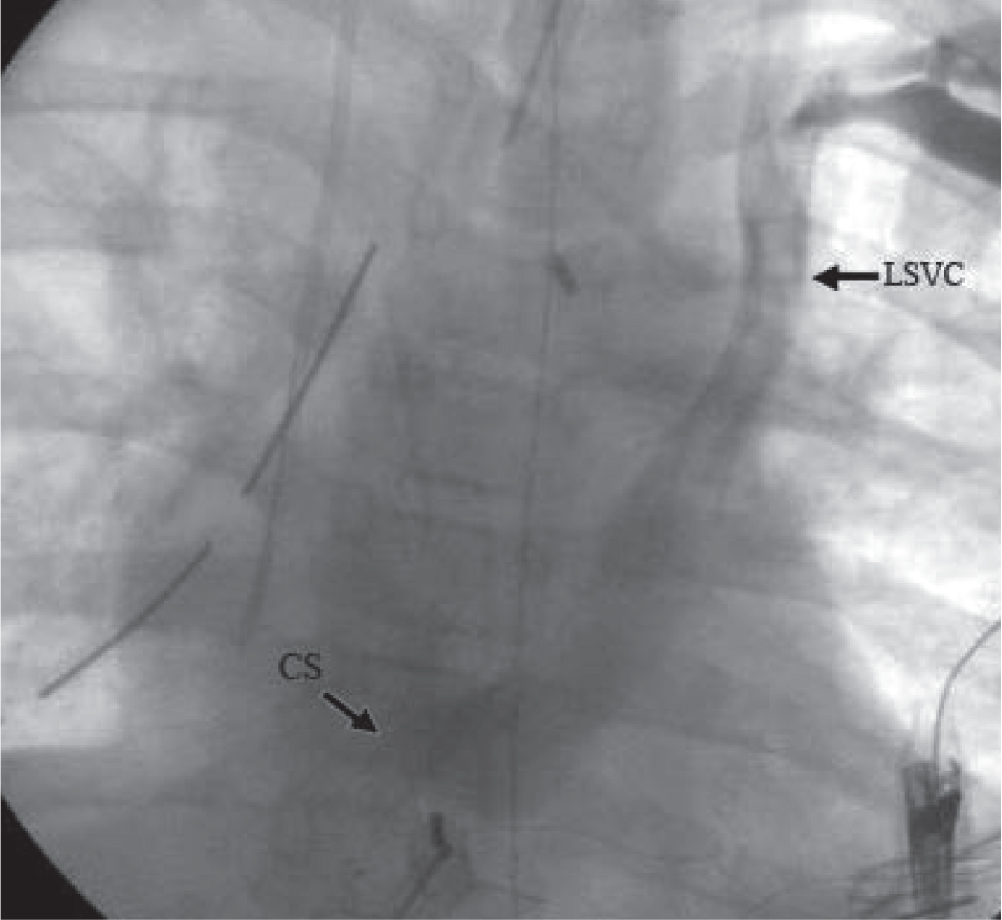
Catheterization study showing LSVC opening to CS
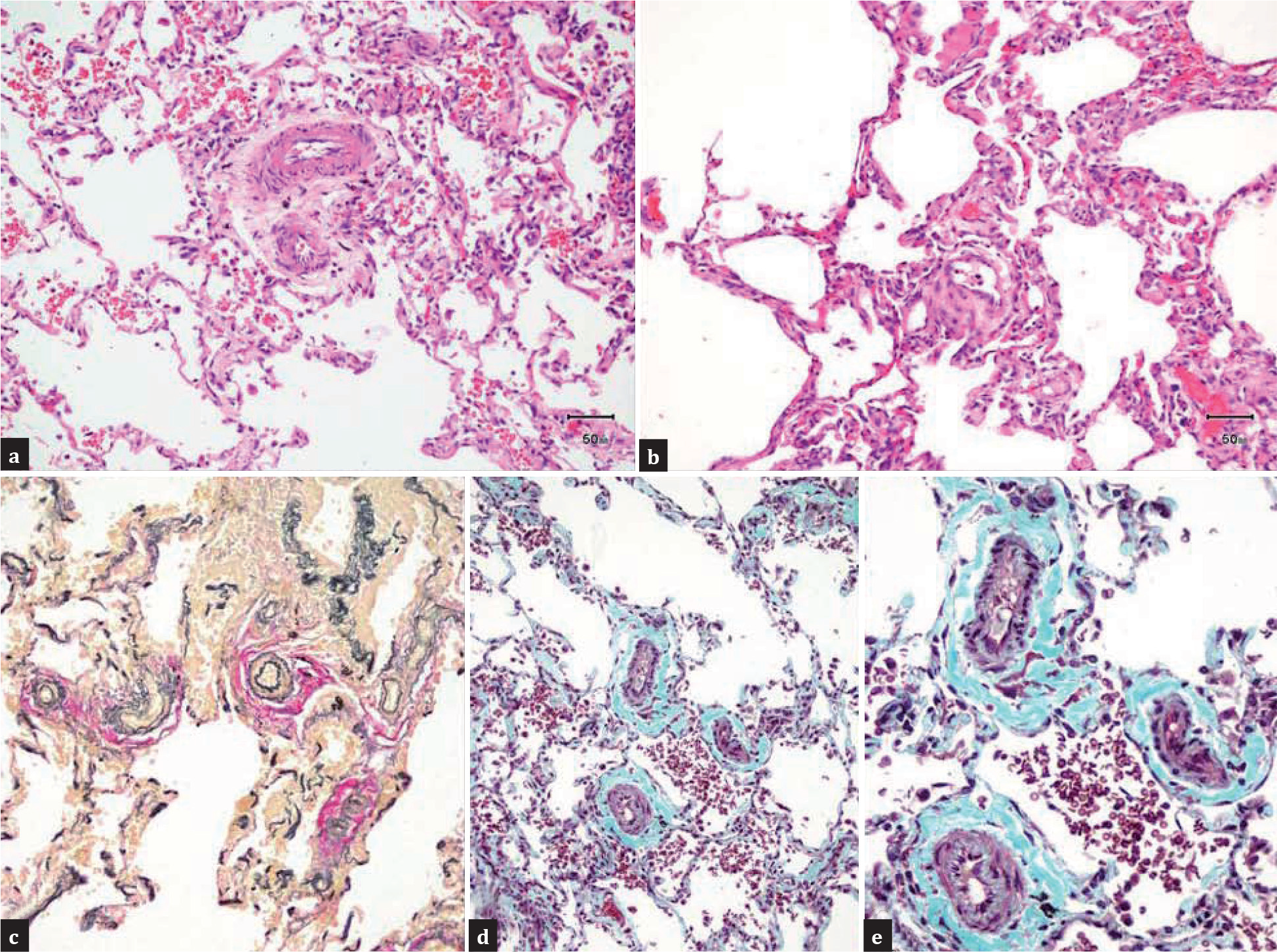
(a and b) Histological section showing prominence of the intracinar arterioles with medial hypertrophy corresponding to Grade I pulmonary hypertension (Heath-Edwards grading system), (H and E x100 (a), x200 (b). (c) Elastic von-Gieson's (EVG) stain highlighting the prominent intracinar arterioles (EVG × 100). (d and e) Muscle hypertrophy along with proliferation of intimal cells in the arterioles corresponding to Grade II pulmonary hypertension (Masson's trichrome x100 (d), x400 (e)
DISCUSSION
The poor clinical results following ICR of TOF are usually due to incomplete correction of the defect. It could be failure to relieve pulmonary stenosis, creation of free pulmonary insufficiency, inadequate closure of VSD, undetected branch pulmonary artery stenosis or absent left pulmonary artery and pre-existing pulmonary hypertension or left ventricular failure.
In the present case, the poor outcome was due to severe PAH together with the development of low cardiac output syndrome, as suggested by the postoperative catheterization study.
Severe PAH following correction of TOF is rare, with a prevalence of about 1%.[1] In some isolated cases, it may be due to previously undetected pulmonary agenesis, pulmonary thrombosis, inadequate closure of the ventricular septal defect, with relief of the pulmonic stenosis, or too large a shunt from a previous Blalock, or, more likely, a Pott's anastomosis.[2–4] In the present case pressure measurement on the operating table, after coming off CPB, showed high RV (55 / 4 mmHg) and PA (48 / 16 mmHg) pressure, with a corresponding systemic pressure of 65 / 44 mmHg. It was accepted, as there was no significant gradient across the RVOT, and the PA pressure was thought to settle in due course. Furthermore, a search for the cause of persistent severe PAH with a postoperative echo and catheterization study showed only mild RPA origin stenosis, which did not explain the finding of severe pulmonary hypertension. However, our patient had DS with a small PDA, which might have contributed to the development of PAH.
Children with DS are at an increased risk of developing PAH due to multiple factors: Congenital heart disease (CHD) with persistent left-to-right shunts, chronic upper airway obstruction, abnormal pulmonary vasculature growth, alveolar hypoventilation, recurrent pulmonary infections, thinner media of the pulmonary arterioles, and diminished number of alveoli.[5–9]
An autopsy study in patients with DS has shown a reduction in the alveolar count, persistence of the fetal double capillary network in the lung, and a reduction in the cross-sectional area of the vascular bed.[6] The increased incidence of persistent pulmonary hypertension in DS is also thought to be due to intrinsic factors, such as abnormal production of NO, and less pulmonary vasodilatation response to NO in DS patients versus control.[10,11]
In addition, following the surgical correction of pulmonary stenosis, significant increase in the pulmonary arterial flow might have enhanced the shear stress on the pulmonary endothelium, causing increased biosynthesis and release of circulating endothelin-1 (ET-1).[12] The elevated plasma levels of ET-1 might be responsible for aggravation of the pre-existing pulmonary hypertension.[13]
A low cardiac output syndrome may be the cause for development of pulmonary hypertension, or, it may occur as a sequel of pulmonary hypertension. Often it is difficult to differentiate between cause and effect. Postoperative low cardiac output may be due to inadequate myocardial protection, ventricle compromised by surgical incision, residual RVOT obstruction, and free pulmonary regurgitation, especially in presence of PAH or residual branch pulmonary stenosis. In a minority of patients it may occur despite a good surgical repair and preserved biventricular systolic function. In such a case echocardiography may show evidence of the restrictive physiology. [14]However, it is a transient phenomenon and usually recovers in 72 hours or so. Persistent LSVC with dilated coronary sinus may also cause left ventricular filling impairment that is difficult to recognize in the postoperative period, and may lead to impaired LV performance.[15]
Post CPB thyroid hormone suppression in children has shown to contribute to the development of postoperative low cardiac output syndrome and PAH. Perioperative thyroid supplement may lead to an improved cardiac output. The aim of the supplement should be to restore the serum T3 level to within the normal limit. The recommended replacement dose of T3 is 0.1 – 0.4 μg/kg/dose at 8 – 12 hour intervals.[16]
Our case developed a low cardiac output syndrome resistant to maximum ionotropic support and administration of thyroid hormone. In such a situation, where there is a failing myocardium unresponsive to ionotrops, providing a period of rest to the myocardium on ECMO may be helpful. This facility was not available to us.
The development of severe PAH due to low cardiac output is unlikely in the present case, as pulmonary artery pressure measurement immediately after coming off CPB also showed severe PAH. However, the persistent postoperative low cardiac output may have contributed to the persistence of PAH and failure of response to the specific pulmonary vasodilator (sildenafil). Inhaled NO, a potent selective pulmonary vasodilator, and its demonstrated synergistic effect with sildenafil, could have been useful, but we could not use it because of its unavailability. The subsequent postmortem lung biopsy with features of medial hypertrophy and intimal proliferation in the intra-acinar arterioles also did not correlate with the recent onset of PAH.
To conclude, severe pulmonary hypertension may occur in a patient with TOF if associated with DS. Development of low cardiac syndrome in these patients can produce a vicious cycle by aggravating the pre-existing PAH, which may lead to a poor outcome.