
Abstract
Pulmonary arterial hypertension (PAH) is a unique disease. Properly speaking, it is not a disease of the lung. It can be seen more as a microvascular disease occurring mainly in the lungs and affecting the heart. At the cellular level, the PAH paradigm is characterized by inflammation, vascular tone imbalance, pulmonary arterial smooth muscle cell proliferation and resistance to apoptosis and the presence of in situ thrombosis. At a clinical level, the aforementioned abnormal vascular properties alter physically the pulmonary circulation and ventilation, which greatly influence the right ventricle function as it highly correlates with disease severity. Consequently, right heart failure remains the principal cause of death within this cohort of patients. While current treatment modestly improve patients' conditions, none of them are curative and, as of today, new therapies are lacking. However, the future holds potential new therapies that might have positive influence on the quality of life of the patient. This article will first review the clinical presentation of the disease and the different molecular pathways implicated in the pathobiology of PAH. The second part will review tomorrow's future putative therapies for PAH.
Keywords
Pulmonary arterial hypertension (PAH) is a severe disease characterized by a progressive increase of pulmonary vascular resistance due to various degrees of adventitia, media, and intima remodeling of the distal pulmonary arteries. This phenotype has been attributed to an increase in pulmonary artery smooth muscle cells (PASMCs) proliferation and resistance to apoptosis.[1] Data from a recent epidemiological study show that 20–50 persons per million suffer from PAH.[23] The underlying causes of PAH were revised during the World Symposium on Pulmonary Hypertension (PH] held in Dana Point, California, USA in 2008. Table 1 presents the groups and subgroups of pulmonary hypertension. The clinical definition of PAH includes a mean pulmonary arterial pressure (mPAP) greater than 25 mmHg at rest, but it also includes extended parameters such as a left atrial pressure, estimated by the pulmonary capillary wedge pressure (PAWP), of 15 mmHg or less.[4] Nonetheless, lack of specific symptoms often leads to a late diagnosis and a worsening of the prognostic.[5] Therapies are limited, not curative, and PAH remains associated with a poor long-term prognosis.[6] The first part of this review will focus on the latest concepts explaining the PASMCs proliferative- and apoptosis-resistant phenotype that contributes to distal pulmonary artery remodeling in PAH, while the second part will discuss tomorrow's future putative therapies for PAH.
Updated clinical classification of pulmonary hypertension Dana Point, 2008

Adapted from “Updated clinical classification of pulmonary hypertension,” by G. Simonneau et al., 2009, Journal of the American College of Cardiology, 54 (1 Suppl), p. S43–54. © 2009 by the American College of Cardiology Foundation Adapted with permission
CLINICAL AND FUNDAMENTAL MANIFESTATIONS OF PAH
Abnormal pulmonary circulation
The normal pulmonary circulation works on high blood flow volume, low-pressure system, and low-resistance regulation. Pulmonary vasodilatation and vasoconstriction balance are important mechanisms that modulate PVR to adapt to changes such as an increase in cardiac output (CO), resulting in a minimal increase in mPAP. To maintain this tightly balanced regulation, the healthy pulmonary vasculature presents many differences compared to systemic vasculature composition. The pulmonary arteries' and arterioles' walls are thinner, and their smooth muscular tones lower than those of the systemic vasculature. Structural changes occur in PAH, such as sustained vasoconstriction, abnormal vascular remodeling with an increase in the size (hypertrophy) and number (hyperplasia) of the smooth muscle cells in the media, and the adventitia of peripheral precapillary pulmonary arteries. The unique plexiform lesions formed by endothelial cells (ECs) lead finally to the obliteration of the arterioles and capillaries.[7–10] The presence of a sustained hypoxic state can be a factor which enhances pulmonary vasoconstriction and leads to pulmonary vascular medial hypertrophy. The combination of those pathological features lead to decreased gas exchange efficiency.[10–12] The mPAP is a function of CO and PVR, as illustrated by mPAP = CO × PVR + PAWP. PVR is inversely related to arterial lumen radius (r) (PVR α 1 / r4), as illustrated in Figure 1. Thus, the PVR can be significantly influenced by only small changes in the intra-luminal radius. Although large and small pulmonary arteries can influence the PVR, structural changes in small arterioles contribute to a more important variation in the PVR when compared to changes in the larger pulmonary arteries, as seen in animals and patients suffering from PAH.[10]
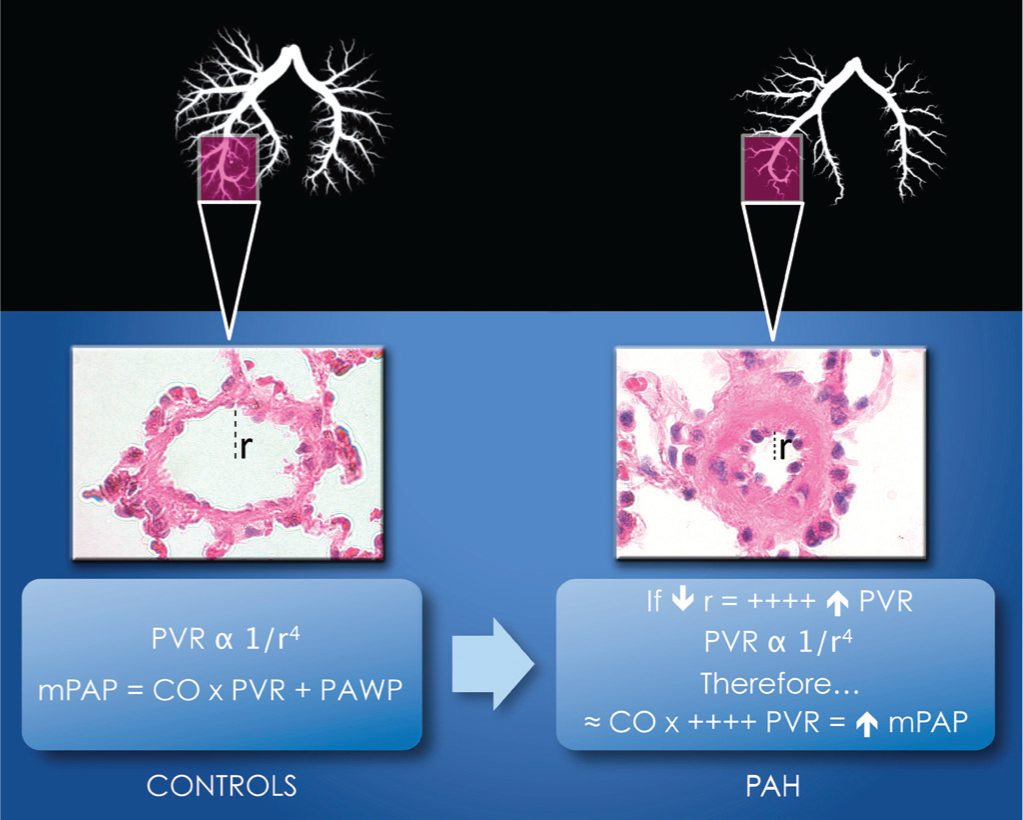
Determinant of abnormal mPAP in PAH. The lumen in control's rat lung allows normal PVR as the artery shows no signs of remodeling. Hence, the mPAP is normal. In the presence of a reduced radius, an increased in the PVR is observed. The function illustrates that only a small change in the radius is needed to elevate the PVR. Therefore, even with a normal CO, the mPAP will be elevated. mPAP = mean pulmonary arterial pressure; r = radius; PVR = pulmonary vascular resistance; CO = cardiac output; PAWP = pulmonary arterial wedge pressure.
Abnormal right ventricular function
The progressive narrowing of the pulmonary microvascular bed, the imbalance between vasodilatation and vasoconstriction, and the presence of in situ thrombosis lead to an increased PVR and mPAP which directly impact the right ventricle (RV). As the RV function declines, a circle of events slowly progress to finally lead to RV failure.[13]
The RV has greater compliance than the left ventricle due to his anatomical particularities, including a thinner wall structure and a crescent shape.[14] The first change due to PAH in RV function is an increase in its afterload, directly related to the increase in PVR and the decrease in pulmonary vascular compliance.[15] The rising systolic, and the following rising diastolic, pressures increase the shear stress applied on the RV wall. Initially, this additional mechanic stress leads to an increase in myocardial mass through an increase in protein synthesis and an increase in cardiomyocyte size by the addition of sarcomere. Therefore, an adaptive RV hypertrophy occurs. In the event of a constant pressure overload, it eventually transits to dilatation. Those changes are well sustained when occurring in the left ventricle, but not in the RV, and the transition toward dilatation/failure occurs much earlier. Although not well understood, evidence shows that it could originate from an imbalance between the oxygen demand and supply in the cardiomyocyte.
Taken together, the increased wall tension and decreased myocardial perfusions progressively lead to a further decreased contractility and dilatation of the RV. As RV function decreases, the increase in RV contraction time and asynchrony leads to a decreased RV stroke volume and by extension to an underfilling of the left ventricle, especially during early diastole.[16–18] The left ventricle filling is also impaired by the progressive development of leftward ventricular septal bowing.[16,17,19] Taken all together, the impaired systolic and diastolic RV function combined with increased mechanical pressure and progressive left ventricle impairment are major components of the reduced cardiac output seen in severe PAH.
SIGNAL INITIATION: CIRCULATING FACTORS
Growth factors
Many growth factors have been identified to play an important role in PAH. When they bind and activate transmembrane cell surface receptor tyrosine kinases (RTKs), they act as potent chemo attractants for cells implicated in PAH pathogenesis like smooth muscle cells, fibroblasts, and ECs.[20]
Vascular endothelial growth factor (VEGF). VEGF has a prosurvival and antiapoptotic role in ECs.[21] Its biological effect is regulated by two RTKs (VEGFR1, VEGFR2).[22] High levels of VEGF[23] and VEGF receptor-2 expressions[24–26] have been observed in PAH patients' lung samples. VEGF and VEGFR2 are expressed by ECs within the plexiform lesions in lungs from patients with PAH.[25,27] Therefore, this angioproliferative growth factor and its receptor have a direct implication in PAH. Short- and long-term exposure to hypoxia upregulates VEGF gene and protein expressions.[28] However, in the monocrotaline (MCT) rat model of PAH, causing an early event of endothelial injury, VEGF expression seems to be decreased.[29] Therefore, VEGF overexpression protects against chronic hypoxia and MCT exposure.[28] A model of severe PAH, caused by inhibition of VEGFR2 expression through SU5416 injection and followed by exposure to chronic hypoxia, has been recently developed. The VEGFR blockade has been shown to cause initial EC apoptosis, followed by the selection of EC clone resistant to apoptosis which, by proliferating, will form severe angio-obliterative lesions contributing to PAH and a right heart failure after exposure to hypoxia.[30]
Platelet-derived growth factor (PDGF) and epidermal growth factor (EGF). Platelet-derived growth factor (PDGF) expression is found in many cell types, including ECs and smooth muscle cells in PAH. It promotes cellular proliferation, migration, survival, and transformation through the activation of two PDGF receptors (PDGFRα and PDGFRβ of RTK family). The expression of PDGF and PDGFR is increased in distal pulmonary arteries of patients with PAH.[31–33] Of particular interest, Schermuly et al. found that PDGFR antagonists effectively treat severe PAH in MCT rats and hypoxic mice by reducing pulmonary remodeling, right heart hypertrophy, and improving CO.[32] Therefore, the inhibition of the PDGFR signalization becomes a potential target to inhibit cellular abnormal proliferation, survival, and migration. Several drugs targeting the PDGFR have been recently investigated in clinical studies with encouraging results.[34,35] A recent study digs the implication of PDGF in cellular proliferation a little deeper. They highlighted the implication PDGF and PDGFR in the Akt/mTor signaling pathway that leads to increased expression of both the stromal interacting molecule (STIMI), a sensor detecting a Ca2+ decrease in the SR/ER membrane, and Orail, a pore forming channel protein in the plasma membrane implicated in the upregulation of the store-operated Ca2+ entry (SO CE), a subsequent rise in the intracellular Ca2+ concentration ([Ca2+].), and resulting in the promotion of PASMC proliferation and tone.[36] Also, Ciuclan et al. proposed a link between PDGFR-β phosphorylation and subsequent enhancement of tryptophan hydroxylase 1 (TPH1) in PAH. Indeed, a hypoxia-dependent TPH1 expression has been showed diminished by administration of PDGFR antagonist.[37] Those new data deepen our knowledge concerning the action of PDGF inhibitors.
The epidermal growth factor (EGF) family had generated a growing interest in PAH. The EGFR/ErbB family includes four receptors, namely the HER1/EGFR (ErbB1), HER/2neu (ErbB2), HER3 (ErbB3), and HER4 (ErbB4). EGFR/ErbB belongs to the RTK family and is highly investigated in many human cancers and the target of several anticancer therapies.[38] An increased EGFR signaling has been associated with EGF-related smooth muscle proliferation and the development of PAH in mice over-expressing TGF-α.[39,40] Thus, the inhibition of EGFR has been thought to be therapeutically interesting. However, the inhibition of this signal only had partial efficacy in MCT rodent models and no significant efficacy on EGFR expression in the lungs of PAH patients.[41]
Pro-inflammatory molecules
The presence of inflammation is commonly observed in the pathological presentation of PAH. In addition, patients with systemic inflammation condition, found in scleroderma or systemic lupus erythematosus, can develop PAH, and generally a more severe form of PAH. Cytokine and chemokine-dependent mechanisms leading to inflammatory cell recruitment are prominent in PAH.[42] Pulmonary vessel infiltration by inflammatory cells like dendritic cells, T and B lymphocytes, and macrophages, as well as increased plasmatic concentrations of interleukin (IL)-1 and IL-6 have been reported in human and experimental PAH, suggesting that inflammation is implicated in the pathophysiology of PAH.[43] Perivascular and interstitial inflammatory infiltrates are predominantly composed of mononuclear cells, mainly lymphocytes, with a minority of macrophages and neutrophils.[27,44] Also, CD44, a cell adhesion molecule, is over expressed in pulmonary artery ECs within plexiform lesions and surrounded by T-cell infiltrates in PAH patients.[45] Recently, the work of Ormiston and colleagues has identified an impairment of natural killer (NK) cells in iPAH, HPAH, and in two widely used animal models.[46] In patients, abnormal NK cell phenotype was marked by a rise in the functionally deficient CD56−/CD16+ subset, not observed in the control group. In animal models, reduction in NK cell number, cytolytic activity, and cytokine production were demonstrated. The innate immunity seems, therefore, to be altered and might play a role in the pathobiology of the disease.
Vascular tone mediators imbalance
ECs produce endothelin, a member of the 21-amino acid peptides family and known to play an important role in vascular tone regulation. Endothelial dysfunctions are observed in PAH, and they lead to a reduced production of vasodilatory mediators.[47,48] In fact, Endothelin-1 (ET-1) expression is increased in PAH compared to healthy subjects, enhancing an increase in intracellular calcium and activating the protein kinase C and resulting in a vasoconstriction of the smooth muscle cells through ETB receptors.[49] Endothelin expression is stimulated by cytokines, catecholamine, or substances from platelet-derived aggregation and can be downregulated by prostaglandins, nitric oxide (NO), and oxidant stress.[50] Moreover, plasma levels of circulating ET-1 correlate with PAH severity.[51] Prostaglandin I2 (prostacyclin) is a strong endogenous vasodilator that increases the activation of cyclic adenosine monophosphate (cAMP) and by this process induces smooth muscle cell relaxation.[52] Additionally, prostacyclin (PGI2) inhibits platelet aggregation and smooth muscle cell proliferation. Prostacyclin is produced in the vascular endothelium using arachidonic acid as a precursor. Prostacyclin production is deficient in ECs of patients with PAH. Indeed, levels of PGI2 metabolites are lower in the urine of PAH patients.[48] Moreover, a decreased production of prostacyclin synthase is seen in PAH small and medium pulmonary arteries.[53] This impaired production of PGI2 leads to vascular tone imbalance and even pulmonary remodeling.
The NO diffuses through vascular smooth muscle cells to convert guanosine triphosphate (GTP) into cyclic guanosine monophosphate (cGMP)[54] that initiates a cascade of reactions that ultimately lead to decreased intracellular calcium ([Ca2+].) level and vascular smooth muscle cell relaxation.[55,56] Thus, the endothelium dependent vasodilatation, relayed by NO, is reduced in hypoxia-induced PAH in rats[57] as well as in human alveolar hypoxia-induced PAH[58–61] and Eisenmenger syndrome-induced PAH.[62] The cGMP concentration also depends on the fifth isoform of the phosphodiesterase (PDE-5) that converts cGMP into GMP, which do not have any effects on vasodilatation.[63] Therefore, reduced NO and cGMP concentrations cannot effectively counterbalance the levels of vasoconstrictive mediators thromboxane and ET-1 that are increased in PAH.[47,48] An elevated plasmatic concentration of serotonin has been identified in severe PAH patients suggesting enhanced serotonin production, transport, and paracrine activity near PASMCs.[64,65] Furthermore, even after heart-lung, single-lung, or double-lung transplantation (the only current way to cure PAH[66]), plasma serotonin concentrations remain increased.[67] Therefore, this persistent abnormality suggests that it originates prior to the development of PAH.
Ions imbalance
Pulmonary vasculature tone is regulated by an important mechanism, the hypoxic pulmonary vasoconstriction (HPV). It is a physiological response of the small pulmonary arteries that diverts deoxygenated blood away from hypoxic alveoli, thus optimizing the matching perfusion and ventilation and preventing arterial hypoxia. Exposition of PASMCs to chronic hypoxia and elevated ET-1 induce structural changes in pulmonary arteries and increase pulmonary vascular tone. Chronic hypoxia reduces the voltage-gated K+ channel (Kv channel) and the two-pore domain K+ channel (K2p) currents, impairs HPV, and reduces O2-sensitive K+ current,[68,69] leading to PASMC membrane depolarization thereby promoting cell growth and migration.[68] More specifically, the sensitivity of K2p channels regulates vascular tone, particularly in small pulmonary vessels.[70–72] The TWIK-related acid sensitive K+ channel K2p 3.1 (TASK1) influences greatly the resting membrane potential in human PASMCs and has been demonstrated to be inhibited by ET-1 in PAH.[72,73] Recently, it has been demonstrated that TASK1 channel inhibition by ET-1 is RhoA and Rho kinase dependent, which contributes to PAH at a molecular level.[74] Also, chronic hypoxia is associated with a decreased expression in messenger ribonucleic acid (mRNA) levels of Kv1.1, Kv1.5, Kv2.1, Kv4.3, and Kv9.3 α-subunits in cultured rat PASMCs.[75–77] Taken together, the change in the Kv and K2p channel currents and the deceased expression of the Kv channels reduce the K+ trans-membrane influx and lead to depolarization.
The increased intracellular calcium ion concentration ([Ca2+].) plays an important role in pulmonary vascular constriction. Chronic hypoxia causes an increased ET-1-mediated vasoconstriction through downregulation of Kv channels, but also an upregulation of the canonical transient receptor potential (TRPC) channel expression, resulting in PASMCs' membrane depolarization and activation of the voltage-operated L-type Ca2+ channels (VOCC), leading to an increased Ca2+ influx.[78,79] In addition, ET-1 increases the sensitivity to Ca2+ in the PASMCs.[80] [Ca2+]i is also increased through voltage independent pathways involving the store-operated Ca2+ channels (SOC) activated by intracellular depletion of Ca2+, and the receptor-operated Ca2+ channels (ROC) activated by interaction of an agonist with a membrane receptor.[81,82] Exposition to chronic hypoxia has been demonstrated to activate the Ca2+-dependant transcription factor Nuclear factor of activated T cells (NEAT) in vitro and in vivo.[83] Increased [Ca2+]i leads to calcineurin activation, which dephosphorylates NFAT and exposes nuclear localization signal that allows NFAT nuclear translocation.[78] Also of interest, the calcium-sensitive potassium channels (BKCa) conduct ionic currents that mediate membrane hyperpolarization and vascular relaxation, and their activities are regulated by membrane potential (Em), [Ca2+]., and channel phosphorylation.[84] It has been demonstrated that BKCa does not modulate HPV in hypoxic condition, but is responsible for hypoxia-induced relaxation due to the resulting increased Ca2+ sensitivity, suggesting that BKCa might play a major role in the regulation of vascular tone in response to hypoxia.[85] Taken all together, chronic PASMCs exposure to hypoxia modifies the ionic movements across the PASMCs, which leads to an increased intracellular K+ and Ca2+ concentration and disrupts tightly regulated mechanisms such as vascular tone and balance between cellular proliferation and apoptosis.
SIGNAL TRANSDUCTION: CRITICAL PATHWAYS THAT MAINTAIN AND AMPLIFY DISEASE PROGRESSION
Disrupted TGF-β signaling
The heritable (HPAH) and idiopathic (iPAH) forms of PAH have been linked with loss-of-function mutations in the bone morphogenetic protein receptor 2 (BMPR2) gene, located on the long arm of the chromosome 2q31–33, and the transforming growth factor β (TGF-β) superfamily of receptors pathways.[86] The bone morphogenetic proteins (BMPs) ligands are essential to cellular proliferation and apoptosis and play a key role in embryogenesis.[87] When the BMP2 ligand binds to BMPR2, it can signal through different pathways, including pSmad1/5,[88] p-p38,[89] pERK, JNK, and Akt/PI3K.[90,91] Mutations in BMPR2 have been reported in 70% or more of HPAH and in approximately 10–20% of patients with sporadic iPAH.[92] However, the penetrance of HPAH remains low with 80% of family members carrying BMPR2 mutations that will never develop PAH.[93] A BMPR2 mutation is rarely seen in patients with PAH related to a congenital left-to-right shunt[94] and is also rarely seen in patients with PAH-associated with appetite suppressants.[95] The functional link between mutations in BMPR2 and PAH is reinforced by the fact that, independent of a mutation in BMPR2, most iPAH patients have reduced BMPR2 protein expression.[96]
Increased ET-1 production has been linked to abnormal BMPR2 signaling; however, it remains to be clearly demonstrated since the interplay and balancing effects between BMPR2 and TGF-β is complex. A recent study presents new elements about this concept by showing that BMPR2 knockdown increases Smad1 and Smad5 phosphorylation and ET-1 production in human lung microvascular ECs.[97] The global gene expression in iPAH is distinct from HPAH and healthy individuals. The authors have related that when not triggered by BMPR2 mutation, iPAH could at least in part possibly originate from increased expression of the transcription factor MSX1—a regulator of BMPs expression—that shows a four-fold higher level in iPAH patients than in healthy controls. Interestingly, MSX1 have been also linked with capillary regression.[98]
Additional genes have been identified as associated with mutations in the effectors of the signaling pathway and the TGF-β superfamily of receptors: Activin receptor-like kinase 1 (ALK1), endoglin (ENG),[99] and more recently identified, Caveolin-1 (CAV1).[100] Mutations in ALK1[101] and ENG[102] have been observed in patients with hereditary hemorrhagic telangiectasia (HHT), an autosomal dominant vascular dysplasia with abnormally dilated vessels forming mucosal and visceral telangiectasia in association with PAH.[101,103] Animal models deepen the implication of ALK1 in the development of PAH as ALK1-deficient mice develop spontaneous pulmonary hypertension. In contrast, BMPR2-heterozygous mice require additional perturbations such as hypoxia and serotonin, or inflammation, to elicit an exaggerated pulmonary hypertensive phenotype[104] indicating a potentially more pathologic effect of ALK1 mutation compared to BMPR2 mutation. Mutations in CAV1 have been assodateci with PAH in a large family with HPAH where six individuals were affected over three generations.[100] Those mutations lead to a decreased formation of caveolae, a subtype of specialized microdomains known as lipid rafts that are rich in cell surface receptors, critical to initiation of a cellular signaling cascade, such as NO pathway and G-protein coupled receptor, and, therefore, important to the homeostasis of the pulmonary vasculature.[100,105]
The STAT3/NFAT pathway
Signal transducer and activator of transcription 3 (STAT3) implication in aberrant PASMC proliferation has been recently highlighted.[106] STAT3 is a transcription factor activated by phosphorylation[107] in response to cytokines (IL-6, TNF),[108] growth factors (PDGF, VEGF),[108] and vasoconstrictor agents (ET-1 and angiotensin 2 [Ang2]).[109] Thus, different types of receptors can be involved in STAT3 signaling such as RTK, G-protein coupled receptors (GPCR), or TNF type receptors. Several downstream STAT3 targets have been identified and the role of STAT3 as a signaling hub has been reinforced. STAT3 not only integrates several upstream signals but also redistributes this signal downstream to enhance several cellular processes to sustain the proproliferative and antiapoptotic phenotype by amplifying the signal. STAT3 has been associated with increased expression of Piml.[110] Enhanced expression of Pim1 promotes the activation of NEAT, which, therefore, promotes inflammation by enhancing cytokine secretion,[111] proliferation of smooth muscle cells by decreasing K+ channels activity which promotes increased [Ca2+].,[112] and apoptosis resistance by increasing the mitochondrial membrane potential. STAT3 also triggers Survivin expression[106,113,114] through activation of the transcription factor Krüppel-like factor 5 (KLF5).[115] Once activated, this zinc-finger-type transcription factor promotes the upregulation of the cyclin B1 and triggers the expression of Survivin, and then contributes to PASMC proliferation and survival. STAT3 is also activated by mechanical stress associated with pressure overload, including shear stress and cyclic strain, through the stimulation of integrin type receptors.[116] Interestingly, STAT3 axis has also been implicated in the downregulation of the endothelial nitric oxide synthase (eNOS] expression and subsequent decrease in NO generation.[117] Indeed, the protein kinase delta (PKCδ) activation following ET-1 stimulation in ovine fetal PAECs has been associated with STAT3 phosphorylation and increased binding of STAT3 on the eNOS promoter, resulting in inhibition of eNOS expression.[117] Thus, STAT3 pathway might be at least in part responsible for the downregulation of the NO signaling in PAH.
Peroxisome proliferator-activated receptor γ
Peroxisome proliferator-activated receptor γ (PPARγ) is a transcription factor well known to be implicated in PAH etiology. It has been suggested that PPARγ is a downstream target of the BMPR signaling pathway.[118] According to the recent literature, PPARγ is involved in a wide range of physiological processes from lipogenesis to inflammation and also plays a critical role in the pathogenesis of PAH because of its effect on proliferation and migration. Hansmann and colleagues demonstrated that the antiproliferative effect of BMP2 was BMPR2/PPARγ/ApoE dependent[119] and that ApoE knockout mice on a high-fat diet develop PAH, which can be reversed by the administration of PPARγ agonist.[120] PPARγ agonist has quickly been considered a possible therapeutic intervention. The activation of PPARγ with insulin-sensitizing agents thiazolidinedione (TZD) in this model help reduce systemic blood pressure, attenuate the formation of arteriosclerotic lesions, and block the arterial wall remodeling process. The use of rosiglitazone allows for the attenuation of pathological arterial remodeling and neomuscularization of arteries in the lung and, therefore, without being able to prevent the development of pulmonary hypertension in rats exposed to chronic hypoxia.[121,122] This drug might have therapeutic application with PAH patients. Questions have been raised about its cardiovascular side effects, but clinical results are still conflicting about its real ratio between risks and efficacy.[123] We recently demonstrated that the receptor of advanced glycation end-products (RAGE) was an upstream target of PPARγ in PAH.[124] Indeed, this protein is implicated in a wide range of pathologies such as cancer, Alzheimer's disease, and vascular diseases.[125–127] From a molecular point of view, RAGE seems to be implicated in many cellular signaling pathways like inflammation, proliferation, and migration,[128,129] all of which are implicated in PAH pathogenesis. Also, RAGE is highly upregulated in PAH lung tissues.[124,130] Thus, we showed that RAGE accounts for STAT3 activation as well as BMPR2 and PPARγ downregulation in PAH-PASMCs, the same phenotype observed when control PASMCs are exposed to S100A4, a RAGE ligand, known to be increased in PAH.[131] Moreover, RAGE inhibition in PAH-PASMCs and in PAH animal models (MCT and Sugen) reverses established PAH, illustrated by decreasing mPAP, vascular remodeling, and RV hypertrophy.
Furthermore, in human pulmonary arterial endothelial cells (PAEC) from PAH patients, a BMPR2-mediated transcriptional complex between PPARγ and β-catenin have been characterized and a disruption of this complex impair BMP-mediated PAEC survival.[119,132] The human PAEC also presents a reduced expression of apelin when compared to PAEC from healthy controls. Apelin have been established to suppress proliferation and to induce apoptosis in PASMC, like BMP-2/BMPR2 ligand complex.[132,133] Cell culture experiments made by Alastalo and colleagues have shown that apelin-deficient PAEC were prone to apoptosis, and were promoting excessive PASMC proliferation.[132] Administration of apelin was able to reverse PAH in mice model where the production of apelin was reduced from the deletion of PPARγ in ECs. Therefore, apelin might represent a therapeutic alternative to treat PAH.
Notch signaling in PAH
Notch signaling is an essential mechanism involved in the cell-fate determination during embryonic development. The Notch receptors and ligands are bound to the cellular membrane. The signal transduction occurs by cell-to-cell contact, leading to a proteolytic cleavage of the Notch receptor, including a final cleavage by the γ-secretase to release the receptor's intracellular domain. The intracellular domain of the Notch receptor translocates to the nucleus to form an active transcription activator complex with the DNA binding protein CBF-1 and activates transcription of HES and HEY genes as its downstream targets.[134] Notch is implicated in many aspects of vascular development like vasculogenesis, angiogenesis, and differentiation of vascular smooth muscle cells.[135] Notch 3 targets gene HES-5, which is expressed exclusively in smooth muscle cells in adults and might be involved in SMCs identity, maturation, and proliferation.[136,138] In vitro PASMCs from PAH patients display higher mRNA and protein levels of Notch 3 and HES-5 than in PASMCs from healthy patients. Overexpression of Notch intracellular domain leads to an increased growth rate of PASMCs, whereas Notch 3-knockout mice lack this proliferative effect.[139] Also, treatment with γ-secretase inhibitor, thus blocking Notch receptor cleavage, attenuates hypoxiainduced PAH in mice. Therefore, these data indicate that high levels of Notch 3 is associated with the development of PAH, favoring a vascular proliferative phenotype.
MicroRNAs: FEED-FORWARD LOOP THAT CLOSES THE SYSTEM
Approximately a decade ago, a 22 nucleotide-non-coding RNA(s) called micro RNA(s) (miR[s]) was discovered in mammalian cells.[140] It now offers us a new vision on post-transcriptional regulatory mechanisms[141] and a new tool to understand physiological and pathological process. Briefly, miRs can inhibit translation by any of the four following mechanisms: (1) Inhibiting translation at a postinitiation step without reducing mRNA abundance, polyadenylation, or polyribsomal content by targeting mRNAs for sequestration in P-bodies; (2) inhibiting translation initiation via a cap-dependent or poly(A)-dependent mechanism, or by affecting physically ribosomal fixation; (3) inducing deadenylation of mRNAs; and (4) inducing direct degradation of the target by other mechanisms.[142–146] There is increasing evidence for the implication of miRNA in many diseases such as cancer,[147] brain disorder,[148] and lung diseases.[149] Interestingly, more than 1,200 miRs have been identified in mammalians cells[150] with, for most of them, a tissue specific expression. Only few miRs have been shown aberrantly expressed in PASMCs and in ECs.
In cancer among others, downregulation of miR-204 has been observed and linked with enhanced cell proliferation and membrane cell depolarization,[151] which are also found in PAH pathogenesis.[83,152] Interestingly, miR-204 downregulation in PAH-PASMCs was found to correlate with PAH severity and to be associated with a higher proliferative rate and resistance to apoptosis.[113] By digging into this concept, and using ChIP-PCR experiments, the authors determined that the downregulation of miR-204 is dependent of STAT3, which binds the promoter of miR-204 host gene TRPM3 and decreases the transcription of the package. miR-204 have been identified to target and downregulate the Src homology 2 domain-containing phosphatase 2 (SHP2), an Src activator; thus when down-regulated, miR-204 enhances a constitutive activation of Src and STAT3. This positive feedback loop involving miR-204 sustains the proproliferative and apoptotis resistant pathologic PASMC behaviour. In addition to the constitutive activation of Src, the downregulation of miR-204 seems to indirectly downregulate BMPR2 by upregulating IL-6 secretion,[153] a potent STAT3 activator, suggesting the existence of another feed-forward loop between miR-204 and STAT3. IL-6 has been demonstrated as upregulated in PAH, although without identifying the cause of this higher secretion.[154] On the other hand, increased IL-6 has been linked to the STAT3-dependant expression of miR-17/92.[155] miR-17/92 has been showed to directly bind BMPR2 and enhance its degradation, suggesting that STAT3 might be responsible for the BMPR2 downregulation. As mentioned above, BMPR2 is involved in the activation of PPARγ which counteracts the STAT3 signaling; however, BMPR2 is also an interacting partner of Src (enhances Src sequestration and decreases its activity). Therefore, STAT3-dependent downregulation of BMPR2 might be associated in two other ways for STAT3 to maintain its own activation. As miRs are also used as biomarker s in oncology, miR-204 could probably endorse this role as it proved to be significantly decreased in the buffy coat of PAH patients.[113,156]
Recent advances in basic science deepen our knowledge about miR-145 andmiR-143. These miRNAs are organized in a cluster regulated by Src and p53 pathways,[157] and they are tightly integrated into a core transcriptional network process involved in smooth muscle differentiation and proliferation. miR-145 appears to direct the smooth muscle cell fate and miR-143 regulates the quiescent and proliferative state of smooth muscle cells.[158] Using a Smad4-mediated transcription, the TGF-β stimulates the expression of the serum response factor and its coactivators, myocardin (Myocd), whereas BMP4 stimulates the myocd-related transcription factor A (MRTF-A) through nuclear translocation. The subsequent upregulation of myocd and MRTF-A induce miR-143 and miR-145 transcription, which repress Krüppel-like factor 4 (KLF4) expression. Therefore, it allows enhanced binding of myocd and MRTF-A to a regulatory DNA element called the CArG box, which promotes contractile gene expression of smooth muscle cells. An unrepressed KLF4 expression reduces excessive binding of myocd and MRTF-A to CArG boxes, which promotes low contractile gene expression of smooth muscle cells.[159] The plexiform lesion and concentric lesions are typically found in severe PAH, and present an abnormal expression of miR-143/miR-145. Plexiform and concentric lesions have increased BMP4 gene expression. Moreover, plexiform lesions have increased TGF-β expression when compared to concentric lesions. The expression of miR-145 was increased in mice exposed to hypoxia and in PAH patients with BMPR2 mutation.[160] miR-143 and miR-145 were significantly higher in concentric lesions compared to plexiform lesions.[161] This points out a possible independent role of miR-145 and miR-143 in characteristic PAH lesions. Their reduced expression in plexiform lesions compared to concentric lesions concurs with the more proliferative profile of this type of lesion. Figure 2 illustrates the integration of miR-204 and miR-143/miR-145 in the pathobiology of PAH.
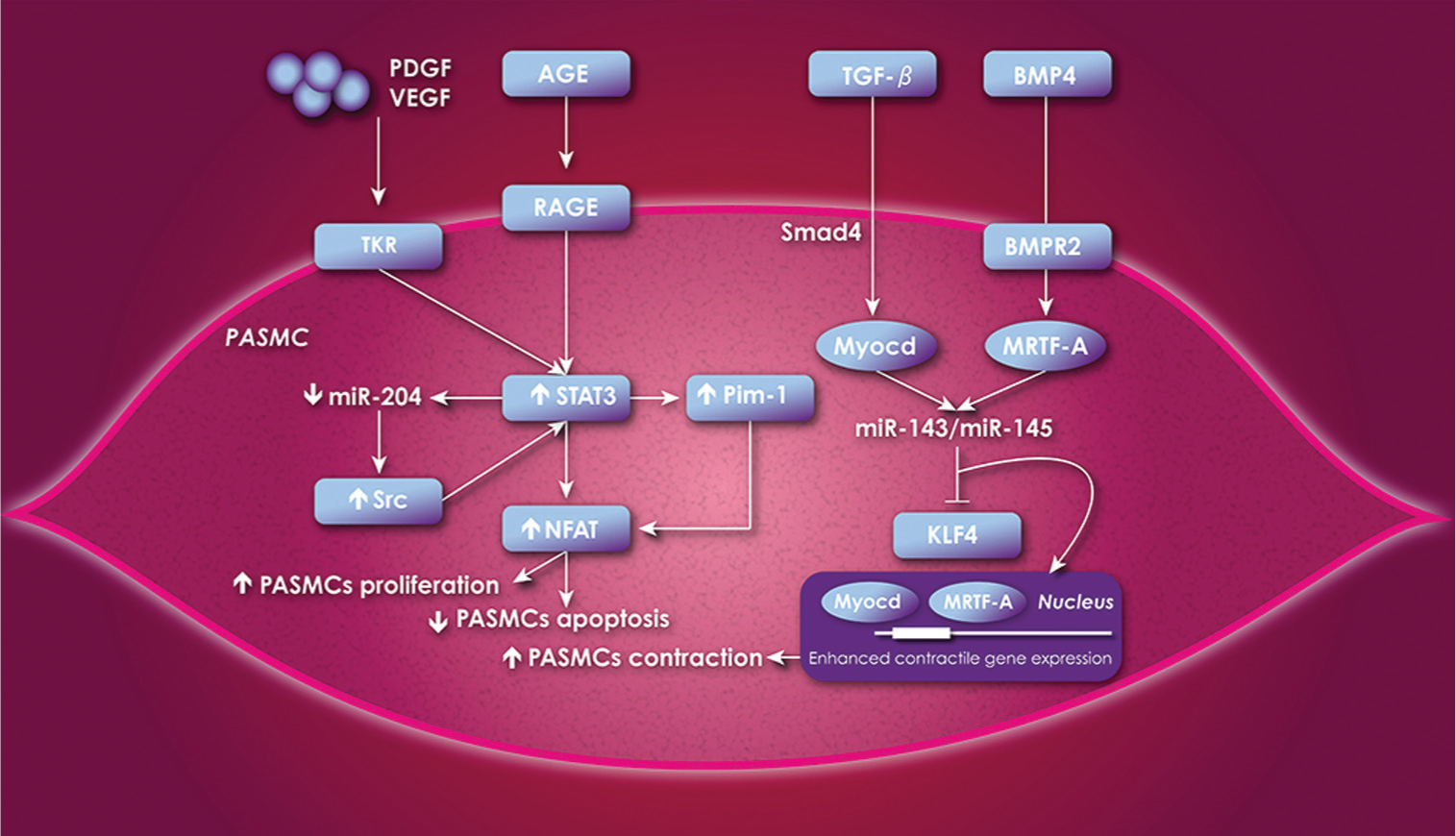
Schematic diagram illustrating the different pathways where miRs are playing an important role in the cellular proliferation and apoptosis of the PASMCs. The increased expression of STAT3 leads to an increased expression STAT3/Pim-1/Src/NFAT axis, which leads to cellular proliferation and reduced apoptosis. TGF-β increases expression of Myocd through Smad4 and BMP4 increases expression of MRTF-A through nuclear translocation, which allow an increased expression of miR-143/miR-145 who then inhibits KLF4. The repressed expression of KLF4 allows an enhanced binding of Myocd and MRTF-A to the CArG box of contractile gene who then promotes PASMCs contraction. If KLF4 is not repressed, there is no enhanced binding of Myocd and MRTF-A to the CArG box of contractile gene, who then presents a low contractile expression. PASMCs = pulmonary arterial smooth muscle cells; VEGF = vascular endothelial growth factor; PDGF = platelet-derived growth factor; AGE = advanced glycation end-product; RAGE = receptor of advanced glycation end-product; Src = sarcoma viral oncogen homolog; STAT3 = transcription factor signal transducer and activator transcription 3; miR = micro-ARN; Pim-1 = proto-oncogene Provirus integration site for Moloney murine leukemia virus; NFAT = nuclear factor of activated T cells; TGF-β = transforming growth factor β; BMP4 = bone morphogenetic protein 4; BMPR2 = bone morphogenetic protein receptor 2; Myocd = myocardin; MRTF-A = Myocd-related transcription factors A; KLF4 = Krüppel-like factor 4.
miR-126 is mainly expressed in ECs and plays a key role in a wide range of physiological and pathologic processes such as cancer,[162,163] brain disorder, regulation of angiogenic signaling, vascular integrity,[164] and recently in PAH, where its expression is deregulated specifically in plexiform lesion.[161] miR-126 is important for vascular integrity, EC proliferation, and neovascularisation.[165] In ECs, its expression is negatively regulated by ET-1 and ET-2 and by the Src pathway. Interestingly, miR-126 has been recently shown to interact with the 3-phosphoinositide-dependent kinase (PI3K/Akt) pathway and the mitogen-activated protein kinase (MAPK) pathway. Its action seems to be indirect, regulating the expression of negative and/or positive regulators such as the Sprouty related EVH1 domain containing protein 1 (SPRED1), phosphoinositide kinase 3 regulatory subunit 2 (PIK3R2), VEGF, insulin receptor substrate 1 (IRSI), or the epidermal growth factor like domain containing protein 7 (EGFL7). Furthermore, mir-126 interacts and regulates the expression of factors involved in apoptosis, modulation of cell cycle arrest, notably by SOX2 and angiogenesis and tumor necrosis factor alpha (TNFα) signaling.[166,167]
Recent reports have identified other miRs as actors in PAH pathobiology. Of interest, plasma miR-150 levels were shown to be reduced and to correlate with survival in PAH.[168] The mechanism by which miR-150 is reduced, however, remains unknown; its expression impairment leads to decreased peripheral number NK cells[169] and B1 cell expansion and to an enhanced humoral immune response.[170] A link could be established with a variety of targets contributing to the deregulation of PASMCs, ECs,[171] and the hypertrophic response of the RV to increased mPAP.[172] Also, miR-150 has been linked with vascular stem cell recruitment, in particular endothelial progenitor cells (EPC), that take active part in PAH pathobiology[173] Recent reports on hypoxia-regulated miRs have identified miR-210 as being the most upregulated by hypoxia,[174–177] butmiR-21 also appears to be highly upregulated by hypoxia, mainly in tumor cell lines,[178,179] suggesting that it may have an important role in cell growth and proliferation. On the other hand, it is interesting to observe that a variety of miRs can be downregulated by hypoxia.[180,181] miR-21 appears to be key regulator of hypoxia responses, linked to abnormal proliferation andmigration of PASMCs.[182] miR-17 impairs EC angiogenic function by targeting the cell cycle inhibitor p21 and the Janus kinase (JAK1) and is upregulated by hypoxia.[183] Treatment with an antagomir-17 has proved its efficacy on MCT rat model by reducing PH, right heart hypertrophy, and by improving the right ventricular function.[184] Moreover, Drakes, Bogaard, and colleagues also demonstrated the implication of miRNAs in RV hypertrophy and failure.[185] This could also provide information on PAH patients' outcomes as RV condition is the main predictor of PAH survival. These oligonucleotides are often the cause of protein misexpression and transcription factor constitutive activation, and are thus great targets to stop inappropriate enrollment of processes that maintain an ongoing progression and worsening of the disease. Thus, miRs are actually seen as a useful diagnostic biomarker and prognostic tool[186] as well as a potential target for future therapeutic interventions.[113] However, more studies are needed to explore the toxicity and risks of miRs therapies on humans.
CRITICAL LACK OF SPECIFIC BIOMARKERS IN PAH
In the current clinical practice, the only used biomarker in PAH remains the N-terminal prohormone of brain natriuretic peptide (NT-pro BNP). It was originally used in the screening and diagnosis of congestive heart failure (CHF), and also to establish CHF prognostic as high NT-pro BNP blood level generally indicates impaired cardiac function and shows poor outcome.[187] The natriuretic peptide does not suit the role of the gold standard biomarker in PAH as it does not reflect changes in the pulmonary vasculature remodeling, but more likely reflects RV stress.[188] Therefore, we don't have biomarkers to detect early stages of the disease and to predict the ongoing vascular remodeling. Current biomarkers are also inefficient to predict the quality and sustainability of the RV adaptive response which deteriorates with the progression of the disease and determines the outcome. Elevated circulating levels of Ang2 were associated with impaired hemodynamics, elevated NT-pro BNP, and unfavorable outcome. Kümpers et al. identified that patients' survival rate was 100% atone year when they had baseline Ang2 < 2.9 ng/mL compared to 78% in patients with baseline Ang2 > 2.9 ng/mL. The patients' survival rate identified during follow-up were 92% versus 63% after two years, 88% versus 54% after three years, and 88% versus 46% after four years, respectively. Therefore, Ang2 seems to have a future as a potential biomarker of severity and outcome for PAH patients.[23] Therapeutic potential of miRNAs should also be confirmed, but they could become important biomarkers, as they are already used as putative diagnostic biomarkers in myocardial infarction, heart failure, coronary artery disease, type 2 diabetes mellitus,[189] and cancer.[190] For PAH, human PAH-PASMCs and cells from the buffy coat of PAH patients have many similarities, as so, they both activate NFAT pathway. miR-204 have been shown to be reduced in PAH patients buffy coat and in extend in PAH-PASMCs, suggesting that miR-204 can become a reliable biomarker.[113] Plasma level of miR-150 also holds promising future,[168] although larger studies are needed. Elevated expression of Pim1 in the buffy coat from PAH patients' makes it an interesting biomarker as well.[106,191] It seems not to be elevated in other inflammatory disease, ruling out PAH patients with scleroderma, a common cause of PAH, increasing at the same time its specificity to PAH. New elements in the clinical toolbox may help to diagnose and treat the disease earlier and thus to improve the outcome drastically.
CURRENT AND FUTURE TREATMENTS IN PAH
Improvement of current therapies in PAH
Endothelin receptor antagonists (ERAs) such as bosentan and ambrisentan were the first orally-administered drugs for PAH treatment and are still currently used in clinics. ERAs are associated to improved cardiopulmonary hemodynamic, exercise capacity and functional class.[192,193] Tissue penetration of bosentan and ambrisentan is limited.[194] However, a tissue-targeting endothelin receptor antagonist called macitentan was recently developed to resolve this issue.[195] A Phase III multi-center study on 743 patients, SERAPHIN, has been initiated to assess the effects of macitentan on mortality and morbidity of macitentan on symptomatic PAH patients. The results of this study are presented in different symposia through the current year. Publication can also be expected throughout the current year.
Treatment with epoprostenol, an intravenous (IV] prostacyclin, is known to improve hemodynamic parameters[196] and symptoms as well as to improve survival in patients with iPAH.[197] However, continuous administration via a catheter is very inconvenient for patients.[6] IV[198] and inhaled tresprostinil[199] and inhaled iloprost[200] are also known to improve hemodynamic parameters. Finally, a Phase II study demonstrated that selexipag, an oral selective prostacyclin receptor agonist, is efficient to reduce pulmonary vascular resistance.[201]
Presently, sildenafil and tadalafil are two PDE-5 inhibitors used for PAH treatment. They both improve exercise capacity, hemodynamic parameters, and quality of life.[202–204] Vardenafil, also a PDE-5 inhibitor (consisting of a different chemical structure) was recently studied in a randomized, double-blind, placebo-controlled study and shows that an oral dose of 5 mg twice daily improves exercise capacity, symptoms, hemodynamics, and clinical outcome.[205] Recently, a novel agent riociguat was developed. Riociguat is an oral soluble guanylate cyclase (sGC) stimulator that transforms guanosine 5′-trisphosphate to cGMP independently of the NO level.[40] Studies on animal models showed that riociguat increases hemodynamic, reduces remodeling, and right heart hypertrophy.[206] A Phase II trial on riociguat in PAH showed improvement in 6-Minute Walk Distance (6MWD) and a decrease in pulmonary vascular resistance.[207] Phase III trials are presently underway and the results are expected soon.
Future therapies in PAH
As previously mentioned, the current treatments are inefficient to improve survival of PAH patients and to reverse the disease. The need of new therapeutic strategies that can cure PAH by acting on remodeling and proliferation is urgent. Similarities found between cancers and PAH, like hyperproliferation, resistance to apoptosis, and cellular metabolism disorders[208] have generated interest for antitumor therapies that may also be applied to the treatment of PAH.
Targeting growth factor. Tyrosine kinase inhibitors, for example, are currently being used against cancer and are now under several clinical trials for PAH. One of them, imatinib, a PDGFR inhibitor, was originally developed for the treatment of different tumors[209] and was tested in several PAH experimental models. It was shown thatimatinib can reverse advanced PAH, decrease right ventricular hypertrophy, and improve cardiac output.[32] In vivo, imatinib has a positive effect on PASMCs by decreasing apoptosis resistance and proliferation.[210] Case studies showed that imatinib can improve the hemodynamic parameters and exercise capacity of severe PAH patient.[35] A Phase II clinical trial was performed on 59 functional Class II-IV PAH patients. The aim of this study was to assess efficacy, tolerability and safely of imatinib in pulmonary arterial hypertension. Patients under imatinib improved significantly their hemodynamic parameters with a decreased PVR and an increase in the cardiac output.[34] However, those results did not reach the primary end-point in 6-MWD with a difference after 24 weeks of +22 m for the imatinib group and −1 m for the placebo group. Moreover, severe side effects including cardiac arrest, syncope, liver dysfunction, and PAH worsening occurred in 39% of the patients under imatinib. The Phase III trials, IMPRES, were initiated to evaluate the efficacy and safely of imatinib in severe PAH in 202 patients. The primary end-point was achieved with a significant change of 32 m in the mean placebo-corrected treatment effect on the 6MWD. Patients under imatinib improved significantly their hemodynamic parameters with significant change in mPAR CO, PVR, and right atrial pressure. However, functional class, time to clinical worsening, and mortality did not differ between treatments.[211] Targeting arterial remodeling by the inhibition of PDGF with imatinib is still considered a potential PAH medication mostly for severe patients, although other studies on liver toxicity and cardiotoxicity need to be conducted.[212] Nilotinib is an active tyrosine kinase inhibitor that was also developed for treatment of chronic myelogenous leukemia.[192] Studies on animals suggest that nilotinib is more effective than imatinib since it reduced right heartpressure and hypertrophy. A Phase II study is underway to assess efficacy, tolerability, and safely of nilotinib on 66 functional Classes II-IV PAH patients. The primary end-point in this case is a change in PVR. Even if nilotinib tends to be safer than imatinib, this potential therapy still represents a risk of cardiac complications and sudden death.[6] Sorafenib is a multikinase inhibitor used for the treatment of renal cell carcinoma and hepatocellular carcinoma. Sorafenib inhibits tyrosine kinases PDGF and VEGF receptor and also serine/threonine kinases including RAF-1, which are related to myocardium hypertrophy. A study on monocrotaline rats showed that sorafenib can reverse experimental pulmonary hypertension and decrease myocardial hypertrophy.[195] A preliminary study on 12 PAH patients was initiated to assess the safely of sorafenib in PAH. This study showed that 200 mg twice daily of sorafenib is well-tolerated by PAH patients and results in an increase in exercise capacity. Moderate skin reactions, diarrhea, and alopecia were the most common adverse events caused by the treatment.[213] However, studies on more patients need to be assessed.
Targeting STAT3. Dehydroepiandrosterone (DHEA] is a hormone known for its vasodilator properties by acting on K+ channels.[214] However, studies on animal models showed that DHEA has an antiproliferative and proapoptotic effect on pulmonary arteries. In fact, Paulin and colleagues demonstrated that DHEA can inhibit the Src/STAT3 axis and by this process improve PAH and decrease RV hypertrophy in monocrotaline rats and human PASMCs.[153] In a recent study, eight patients with pulmonary hypertension associated with chronic obstructive pulmonary disease were daily treated with 200 mg of DHEA. After three months, all patients showed an improved 6MWD and pulmonary hemodynamic without changes in their gas exchange parameters.[215]
Plumbagin (PLB], or 5-hydroxy-2-methyl-1, 4-naphthoquinone, is an organic compound known to have anticancer properties on different tumor cells.[216] A study on nonsmall cell lung cancer (NSCLC) culture has demonstrated that PLB has apoptotic and antiproliferative effects on NSCLC by the downregulating Survivin and growth factor receptor expression.[217] Moreover, another study on myeloma cells showed that PLB suppresses STAT3 activation which is implicated in PAH remodeling.[218] An in vitro study on human PASMCs presented that plumbagin can inhibit PASMC proliferation and resistance to apoptosis by blocking the activation of STAT3/NFAT. Also, it was demonstrated in vivo that oral administration of plumbagin reduced mPAP, vascular remodeling, and right ventricular hypertrophy on sugen and MCT rats.[219]
Circulating progenitor cells. The blood vessels form a complex network of arteries, capillaries, and veins designed to transport micromolecules, macromolecules, gases, and fluids to every cell in the organism. The formation of this network goes through several stages during embryonic development and postnatal development, including vasculogenesis and angiogenesis. Vascular remodeling is needed for the development of a strong network able to sustain shear stress while being able to transport oxygen and nutriments to the physiological environment. We explored earlier the mechanisms implicating VEGF and VEGFR2 expression and leading to endothelial dysfunctions. Proangiogenic factors also have a role in the abnormal vascular remodeling observed in PAH. Defective mobilization and recruitment of endothelial progenitor cells (EPCs] have been implicated in the sustainability of the PAH phenotype.[220] Patients with Eisenmenger syndrome and IPAH have a reduced number of EPCs.[221,222] However, investigators have also found an increased number of EPCs in patients with other forms of PAH.[223,224] Thus, despite the presence of EPCs, there is no successful repair of the damaged pulmonary endothelium. Additionally, an increased expression of progenitor cell markers has been observed in lung tissue from patients with IPAH compared with control lung tissue.[222] Angiopoietin are angiogenic factors essential to vascular development and maturation. Ang1 and its antagonist Ang2 are needed to bind the extracellular domain of the tyrosine kinase receptor Tie-2, which is mainly expressed in ECs. Ang1 stabilize the development of newly formed blood vessels by recruiting mural cells and promoting the maturation and structure integrity of the vessels. In contrast, the presence of Ang2 destabilizes the vessels in the presence of VEGF or promotes vascular regression and ECs death in the absence of VEGF. In patients with iPAH, all angiogenic factors were elevated while histological studies showed that Ang2 mRNA expression was over-expressed in plexiform lesions from IPAH patients' lung tissue.[23]
CONCLUSION AND FUTURE DIRECTION
In the past five years, the understanding of molecular origins of pulmonary arterial hypertension has given rise to numerous research and important discoveries. In fact, the increased knowledge in pathogenesis and the presentation of promising therapy in PAH such as miRNAs, tumor suppressors, and oncoprotein inhibitors provide significant hope to cure this fatal disease. As studies targeting vascular remodeling using antiproliferative agents obtained great success in experimental models, and since many other studies are presently underway, it is allowable to believe that the next decades will be determinant to cure PAH.