
Abstract
Reactive oxygen species (ROS) have emerged as critical players in the pathophysiology of pulmonary disorders and diseases. Earlier, we have demonstrated that ROS stimulate lung endothelial cell (EC) phospholipase D (PLD) that generates phosphatidic acid (PA), a second messenger involved in signal transduction. In the current study, we investigated the role of PLD signaling in the ROS-induced lung vascular EC barrier dysfunction. Our results demonstrated that hydrogen peroxide (H2O2), a typical physiological ROS, induced PLD activation and altered the barrier function in bovine pulmonary artery ECs (BPAECs). 1-Butanol, the quencher of PLD, generated PA leading to the formation of physiologically inactive phosphatidyl butanol but not its biologically inactive analog, 2-butanol, blocked the H2O2-mediated barrier dysfunction. Furthermore, cell permeable C2 ceramide, an inhibitor of PLD but not the C2 dihydroceramide, attenuated the H2O2-induced PLD activation and enhancement of paracellular permeability of Evans blue conjugated albumin across the BPAEC monolayers. In addition, transfection of BPAECs with adenoviral constructs of hPLD1 and mPLD2 mutants attenuated the H2O2-induced barrier dysfunction, cytoskeletal reorganization and distribution of focal adhesion proteins. For the first time, this study demonstrated that the PLD-generated intracellular bioactive lipid signal mediator, PA, played a critical role in the ROS-induced barrier dysfunction in lung vascular ECs. This study also underscores the importance of PLD signaling in vascular leak and associated tissue injury in the etiology of lung diseases among critically ill patients encountering oxygen toxicity and excess ROS production during ventilator-assisted breathing.
Keywords
Phospholipase D (PLD), a ubiquitous enzyme present in fungi, bacteria, plants and mammals, hydrolyzes the membrane phospholipid, phosphatidylcholine (PC) to phosphatidic acid (PA) and choline.[1–3] Among the six isoforms of PLD, PLD1 and PLD2 have been well-characterized. The physiological significance of PLD1 and PLD2 activation is not well-understood. Nevertheless, the activation of PLD1 and PLD2 has been implicated in mitogenesis, cytoskeletal reorganization, protein trafficking and secretion.[2] PA generated by the PLD pathway has been shown to act as a second messenger in mammalian cells. Studies have shown that exogenous addition of PA activates 1-phosphatidylinositol 4-kinase (PI4kinase), mitogen-activated protein kinases (MAPKs), protein kinase C (PKC), NADPH oxidase and alters the intracellular free calcium levels.[2,4] Also, PA is converted to either diacylglycerol (DAG) or lyso-PA (LPA) by lipid phosphate phosphatase or phospholipase A1/A2, respectively.[2] While DAG is an endogenous activator of certain protein kinase C (PKC) isoforms,[1,2,5] LPA transduces signals by binding to the G-protein-coupled LPA receptors.[3] Earlier, others and we have demonstrated that a wide variety of agonists and reactive oxygen species (ROS) activate PLD in the vascular endothelial cells (ECs).[4,6–16] Furthermore, many of the agonists that activate PLD also have been shown to alter the EC barrier function.[2] Recent studies have revealed that exogenous PA alters the paracellular permeability in ECs[17] suggesting a role for PLD in the EC barrier dysfunction. ROS play an important role in cell signaling pathways[18–20] and are implicated in the pathophysiology of a number of lung and cardiovascular disorders.[21] Our earlier studies have demonstrated that ROS activate PLD and modulate the distribution of cytoskeletal and focal adhesion proteins in bovine pulmonary artery ECs (BRAECs).[4,6–8,22–24] However, a direct connection between the ROS-induced PLD activation and lung vascular EC barrier dysfunction has not been established to date. To further establish a physiological role for the intracellularly generated PA, in the current study, we investigated the ROS-induced PLD activation and barrier dysfunction in lung vascular ECs. The results of this study demonstrated that the PLD-generated intracellular bioactive lipid signal mediator, RA, was an essential player in the ROS-induced barrier dysfunction in lung vascular ECs.
MATERIALS AND METHODS
Cell culture
Bovine pulmonary artery ECs (BRAECs), passage number 16 (American Type Culture Collection, Manassas, VA) were cultured in the minimal essential medium (MEM) containing 10% fetal bovine serum (FBS), antibiotics, non-essential amino acids and 5 μg/ml of endothelial cell growth supplement. Cells were cultured in T-75 flasks in a humidified atmosphere of 5% CO2 and 95% air at 37o C. Confluent cells were treated with 0.025% trypsin containing 0.02% EDTA and plated onto sterile 35-mm dishes or on membrane inserts or gold electrodes for PLD activation, cytoskeletal reorganization and barrier function studies.
Assay of phospholipase D activation in intact cells
BPAECs cultured in 35-mm dishes were labeled with [32P]-orthophosphate (5 Ci/ml) in DMEM phosphate-free medium containing 0% FBS for 18–24 hours.[15,16] The radioactive medium was aspirated and the cells were stimulated with vehicle or agonist in DMEM medium containing either 0.05% 1-butanol or 2-butanol for varying time periods as indicated. The incubations were terminated by addition of methanol: HCl (100:1 v/v) and lipids were extracted.[15,16] [32P]-Labeled phosphatidylbutanol (PBt) formed, due to PLD activation and transphosphatidylation of PA to 1-butanol, but not 2-butanol, was separated by thin-layer chromatography on 1% potassium oxalate impregnated silica gel H thin layer plates using the upper phase of ethyl acetate: isooctane: glac.acetic acid: water (65:10:15:50 by vol.) as the developing solvent system. Radioactivity associated with PBt was quantified by liquid scintillation counting and data were expressed as dpm normalized the 106 counts in total lipid extracts or as % control.
Assay of paracellular permeability of albumin across ec monolayer
Permeability of albumin, conjugated to Evans blue, across the BRAEC monolayers was carried out essentially as described earlier.[25] The EC paracellular permeability assay trans-well chamber consisted of sterile plastic wells divided into an upper (luminal) and lower (abluminal) compartments being separated by a membrane filter (Nucleopine, Pleasantus, Calif., USA) on which BPAECs were seeded to confluence. Albumin transport across the monolayer was determined by measuring the absorption of Evans blue dye in medium of the abluminal chamber at 620 nm in a plate reader (Molecular devices, Menlo Park, Calif., USA). Albumin clearance rates were compiled by linear regression analysis of control and experimental groups.
Transendothelial cell electrical resistance measurement
Transendothelial cell electrical resistance (TER) was measured with an electrical cell impedance sensor (ECIS) system[26] (Applied Biophys., Troy, N.Y., USA). The total electrical resistance across the monolayers was composed of the resistance generalized between the basal surface of the cell and the electrode as well as by the resistance between the cells. A 4,000 Hz AC signal with 1-V amplitude was applied to the cells through a 1 MH resistor, creating an approximate constant current source (1 in A). The lock-in amplifier attached to the electrodes detected the changes in both the magnitude and phase of the voltage appearing across the ECs. Resistance data were normalized to the initial voltage and plotted as normalized resistance by utilizing the manufacturer's software.
Preparation of cell lysates and immunoblotting
After challenging BRAECs with the agonists, cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to the polyvinylidene fluoride (PVDF) membrane (30 V for 24 hours), blocked with 5% bovine serum albumin (BSA), incubated with the primary antibodies (1:1,000 dilution), washed and exposed to the secondary antibodies (1:2,000 dilution) as described earlier.[23]
Antibodies
Antibodies for PLD1 (Cell Signaling Technology, Danvers, Mass., USA), Actin (Sigma, St. Louis, Mo., USA), VE-Cadherin (BD Biosciences, San Jose, Calif., USA), Alexa Fluor 568 Phalloidin and Alexa Fluor 488 (Invitrogen-Molecular Probes, Eugene, Oreg., USA) were all commercial purchases. Antibody for PLD 2 was provided by Dr. Nozawa and Dr. Banno (Gifu International Institute of Biotechnology, Japan).
Infection of BPAECs with adenoviral hPLD1 and mPLD2 wild type and dominant-negative mutants
BPAECs grown in 35 mm dishes to ~80% confluence in complete medium were infected with the adenoviral constructs (5 p.ff.u./cell) of Vector-control, PLD1 wild type (Wt), PLD1 dominant-negative mutant (Mn), PLD2 Wt, and PLD2 Mn as described earlier.[23]
Immunofluorescence microscopy
BPAECs grown on plastic slide chambers were treated with ROS or agonists and VE-cadherin and actin redistribution was determined by immunofluorescence microscopy.[23] Slides were examined with a Nikon Eclipse TE 2000-S fluorescence microscope and Hamamatsu digital camera (Japan) using a 60x oil-immersion objective lens and MetaVue software (Universal Imaging Corp., PA).
Statistical analysis
Albumin clearance rates in individual wells were analyzed by linear regression using the Epistat 2.0 public domain software and slopes were averaged from at least six individual determinations. Paired t-test was used to compare pretreatment and post-treatment slopes within the same control or EC chamber. ANOVA with Student-Newman-Keuls test was used to compare mean of clearance rates of two or more treatment groups. Differences were considered significant at P > 0.05 unless otherwise stated. Data are expressed as mean ± S.D.
RESULTS
Reactive oxygen species induces phospholipase D activation and barrier dysfunction in endothelial cells
In order to establish a plausible link between the ROS-induced PLD activation as a pre requisite for the permeability alterations, BPAECs were exposed to H2O2 (100 μM) for 30 minutes and PLD activation was determined by determining the extent of [32P]-PBt formation. In parallel experiments, BPAECs were exposed to H2O2 (100 μM) for 180 minutes and the paracellular transport of Evans blue-conjugated albumin across the EC monolayer was determined. As shown in Figure 1A, the formation of [32P]-PBt, an index of PLD activation, was rapid and observed 30 min after the addition of the agonist. In contrast to PLD activation, increase of albumin clearance in response to H2O2 exposure occurred at later time periods between 60 and 180 minutes of exposure (Fig. 1B). These results demonstrated that H2O2 mediated PLD activation preceded the barrier dysfunction in BPAECs.
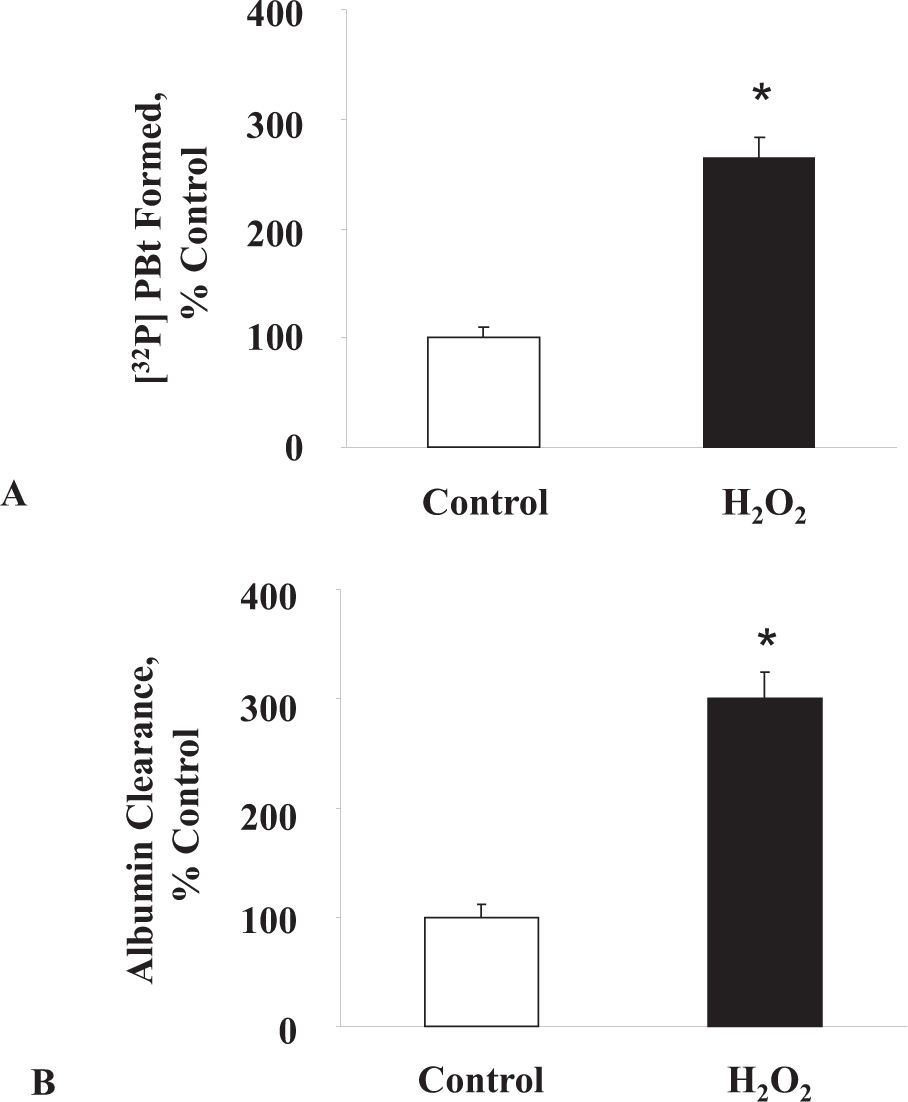
H2O2 activates PLD and induces albumin clearance in ECs. BPAECs were grown on sterile 60-mm dishes or plastic inserts and PLD activation and albumin flux following H2O2 (100 μM, 30 min) treatment were measured as described under “Materials and Methods”. Values are mean ± S.D of three independent experiments in triplicate. *Significantly different from control untreated cells (P > 0.05).
1-butanol, but not 2-butanol, attenuates ROS-induced PLD activation and EC paracellular albumin permeability
The above results suggested a potential link between the agonist-mediated PLD activation and barrier dysfunction in ECs. So as to demonstrate a role for PLD in the oxidant-induced EC barrier dysfunction, we used the primary alcohol such as 1-butanol, the preferred substrate of PLD-catalyzed transphosphatidylation reaction and the secondary alcohol, 2-butanol, which does not act as a PLD substrate. Through the transphosphatidylation reaction, PLD utilizes 1-butanol and converts PA into phosphatidylbutanol (PBt) at the expense of PA and renders physiologically active PA into inactive PBt.[2] If the barrier dysfunction induced by ROS were mediated by the PLD pathway, transphosphatidylation of PA in presence of the primary alcohol (1-butanol) to PBt would be expected to decrease the paracellular transport of albumin across the EC monolayer more effectively than the secondary alcohol, 2-butanol, which is not the preferred substrate of PLD. Hence, (Fig. 2A) shows the effects of 1-butanol and 2-butanol on the formation of [32P]-PA and [32P]-PBt by H2O2 in BPAECs. In the presence of 1-butanol, but not 2-butanol, [32P]-PA generated by PLD activation was efficiently converted to [32P]-PBt. Identical concentrations of 1-butanol, but not 2-butanol, caused the attenuation of H2O2-induced barrier dysfunction as measured by the albumin flux across the BPAEC monolayer (Fig. 2B). These results demonstrated that PA generated by PLD activation regulated the ROS-induced barrier dysfunction in ECs.
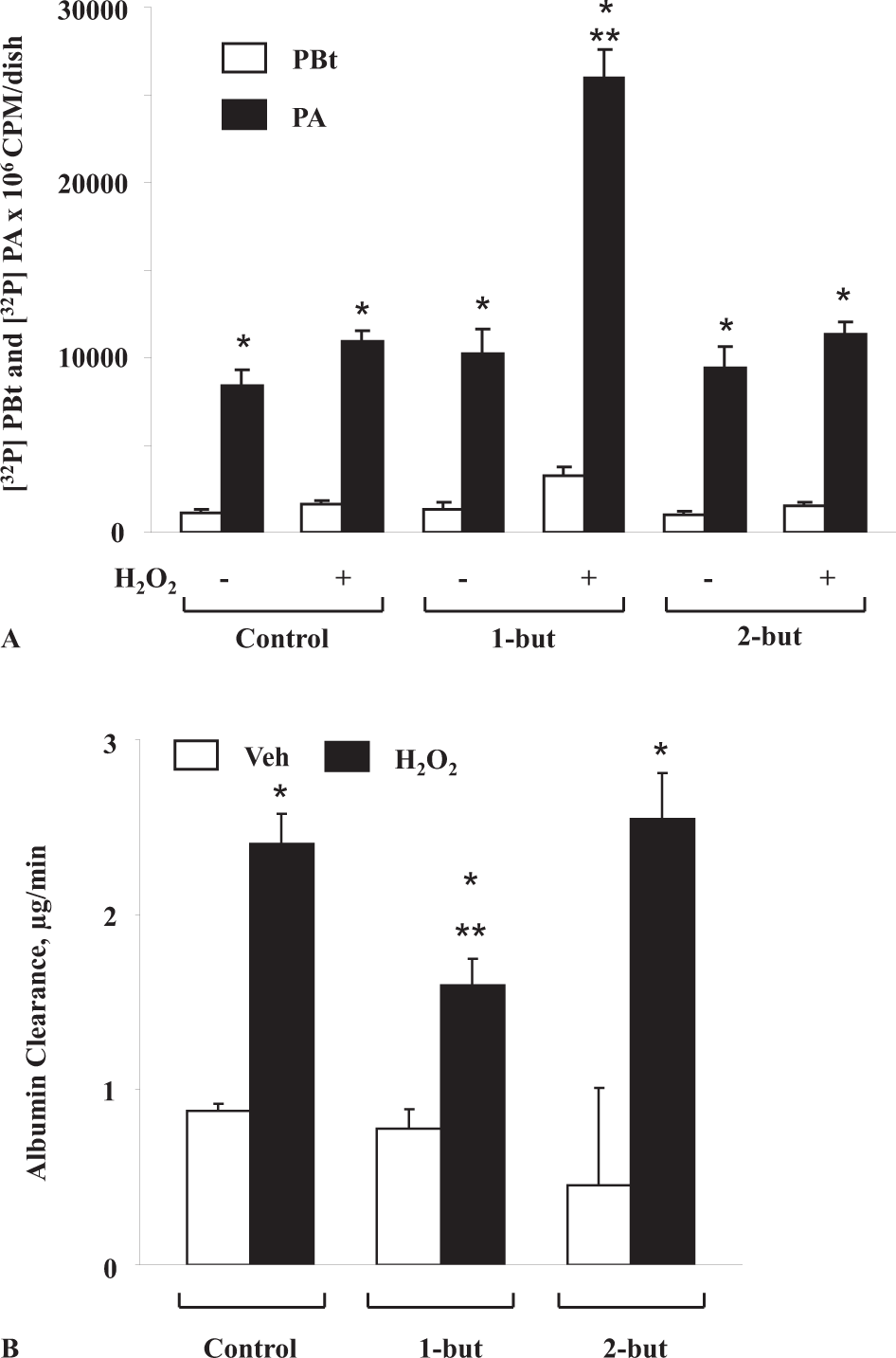
PA generated by PLD pathway regulates oxidant-induced barrier dysfunction in ECs. BPAECs grown on sterile 60-mm dishes, pretreated with 1-butanol and 2-butanol, stimulated with 100 μM H2O2 for 30 min and transphosphatidylation of PA to PBt
Ceramide attenuates ROS-induced PLD activation and paracellular permeability of albumin in ECs
Earlier studies have shown that C2- and C6- ceramides inhibit the agonist-stimulated PLD activation in mammalian cells.[4] Therefore, with the purpose of further establishing the role of PLD in the ECs paracellular transport of macromolecules, here we investigated the efficacies of cell-permeable ceramides on the H2O2-mediated PLD activation and albumin flux across the BPAEC monolayer. The H2O2 -induced PLD activation was attenuated by C2-ceramide (25 μM, 1hour) but not by C2-dihydroceramide (25 μM, 1hour), a physiologically inactive analog of C2-ceramide (Table 1). Similarly, C2-ceramide, but not C2-dihydroceramide, partly blocked the H2O2-induced increase in albumin paracellular transport across the BPAEC monolayer (Table 1). C6-ceramide, in a similar fashion, blocked the PLD activation and albumin paracellular transport induced by H2O2, confirming a role for PLD activation in the ROS induced barrier dysfunction in ECs (data not shown). These results further showed a role for PLD activation in the ROS-mediated endothelial barrier dysfunction.
Effect of C2 ceramide and C2-dihydroceramide on PLD activation and album in clearance

Dominant negative hPLD1 and mPLD2 mutants attenuate ROS-induced paracellular permeability in ECs
ECs have been shown to predominantly express the PLD1 and PLD2 isoforms[2] and to further establish a role of these two PLD isoforms in EC barrier dysfunction, we studied whether over-expression of the Wt hPLD1 and mPLD2 or dominant-negative mutants of hPLD1 or mPLD2 would modulate the H2O2-induced PLD activity and paracellular permeability. BPAECs over-expressing PLD1/PLD2 Wt or catalytically inactive mutants were labeled with [32P]-orthophosphate and exposed to H2O2. In the PLD Wt over-expressing cells, H2O2- mediated [32P]-PBt formation was higher (~2 fold) as compared to the same in vector control, while in the hPLD1-K 898 R/mPLD2-K 758 R mutants, the formation of labeled PBt was attenuated (data not shown). As shown in Figures 3A and B, treatment of ECs with H2O2 decreased the TER that was time-dependent. Transfection ECs with PLD1 and PLD2 catalytically inactive mutants (Figures 3A and B) significantly prevented the ROS-induced barrier dysfunction. These studies further supported a role for PLD1 and PLD2 activation and PA in the ROS-induced barrier dysfunction in the lung vascular ECs.
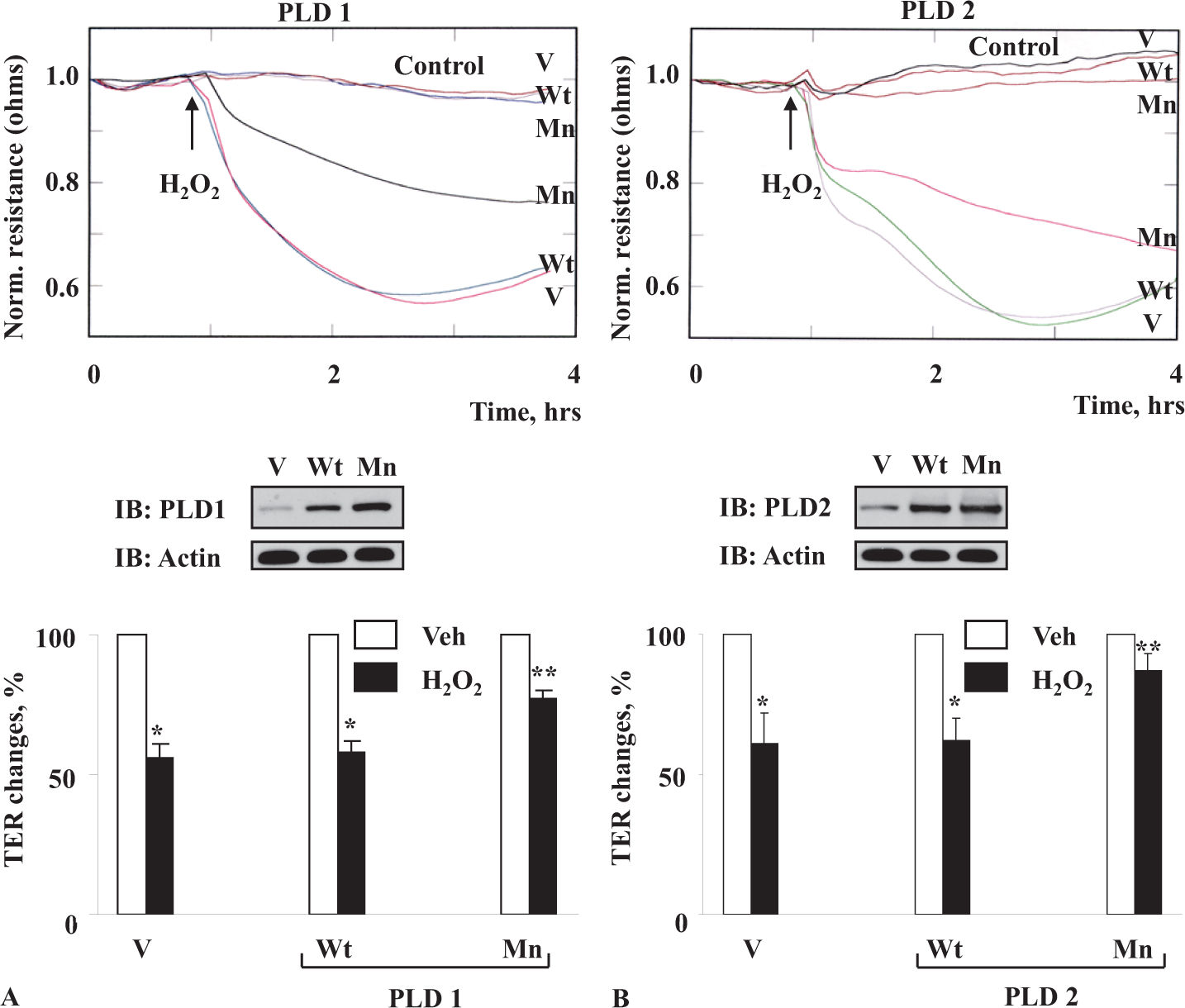
Downregulation of PLD activity attenuates H2O2-induced decrease in TER in ECs. BPAECs grown on gold microelectrodes to ~95 % confluence were infected with vector (v) or wild type (Wt) or mutant (Mn) hPLD1
Role of PLD1 and PLD2 in ROS-mediated cytoskeletal reorganization and focal adhesion protein redistribution in ECs
Our recent study has demonstrated that H2O2 treatment in endothelial cells induces the reorganization of cytoskeleton and focal adhesion proteins and decreases the TER, suggesting that the cytoskeleton and focal adhesion proteins play an important role in the oxidant-mediated vascular endothelial barrier function that is regulated by the cell-cell and cell-matrix contacts.[22,24,27] Treatment of ECs with H2O2 resulted in the formation of actin stress fibers in the vector-control and Wt hPLD1/mPLD2 over-expressing ECs, while the same under identical conditions was attenuated in the hPLD1-K 898 R/mPLD2-K 758 R mutants (Figures 4A and B). Similarly, H2O2 caused the redistribution of VE-cadherin (a focal adhesion protein) to the cell periphery in the ECs transfected with the vector-control and hPLD1/mPLD2 Wt DNA, whereas those responses were attenuated in the cells transfected with hPLD1/2 Mn DNA (Figures 5A and B).
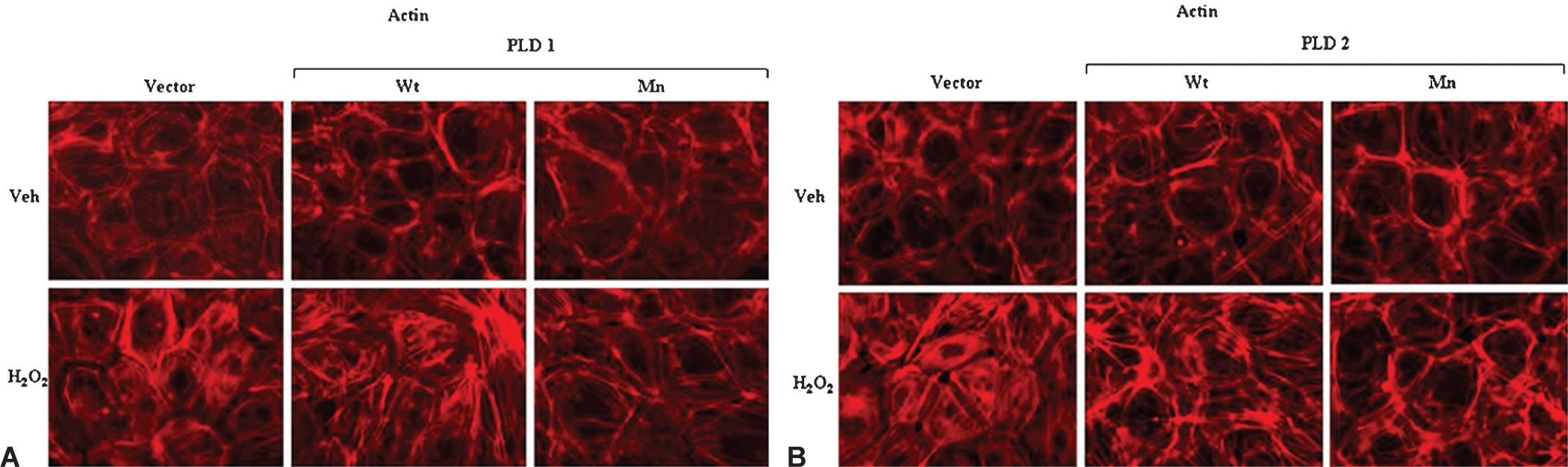
Downregulation of PLD attenuates H2O2-mediated actin cytoskeleton rearrangement in ECs. BPAECs grown on sterile glass coverslipes to ~95% confluence were infected with vector (V) or wild type (Wt) or mutant (Mn) hPLD1
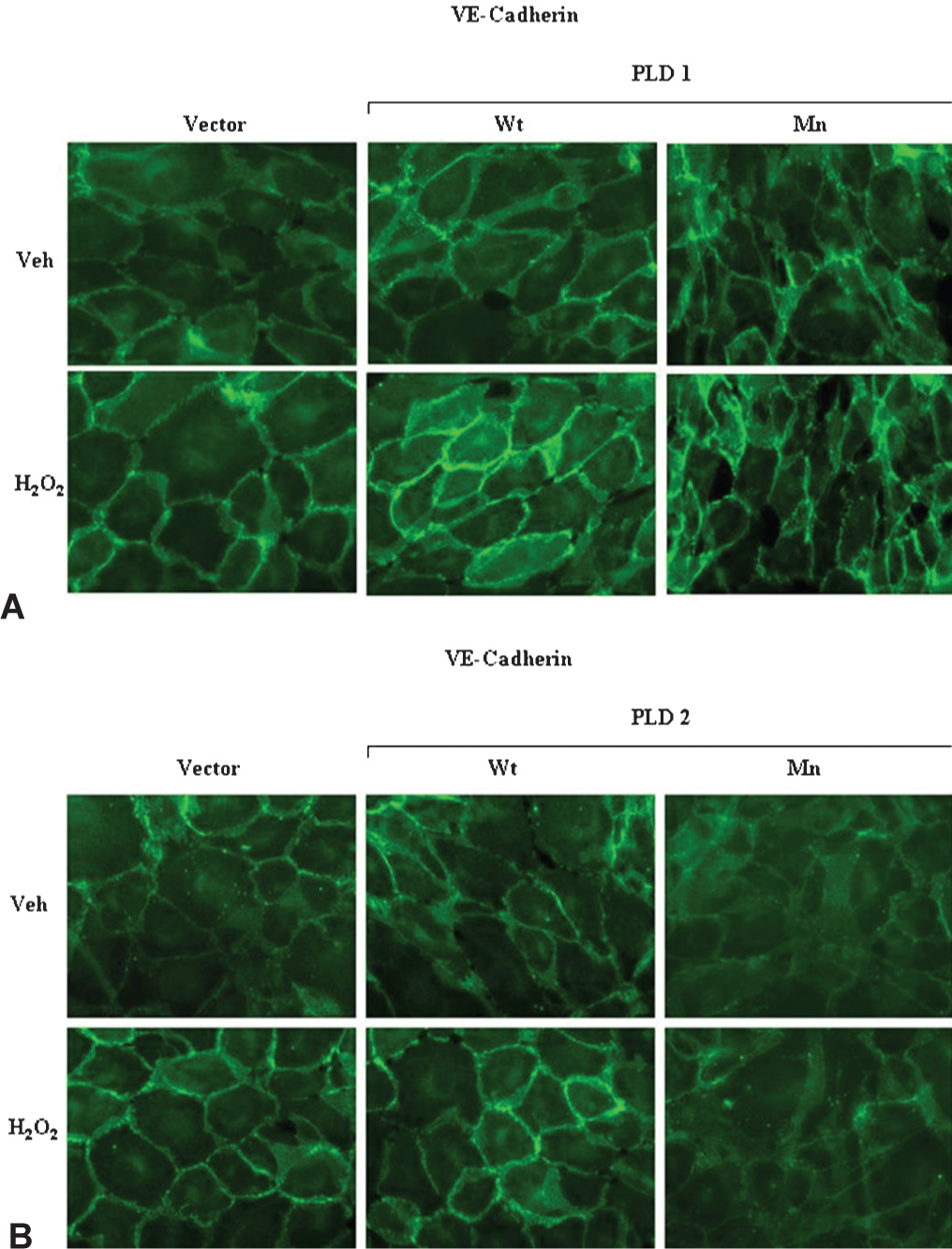
Downregulation of PLD attenuates H2O2-mediated VE-Cadherin redistribution in ECs. BPAECs grown on sterile glass coverslips to ~95 % confluence were infected with vector (V) or wild type (Wt) or mutant (Mn) hPLD1
DISCUSSION
ECs, lining the blood vessels, serve as a semi permeable barrier to circulating cells, plasma albumin, macromolecules and bioactive agents. Maintenance of EC barrier integrity is critical for vessel wall homeostasis and normal organ function.[28] Among various circulating edematic agents, ROS generated at sites of inflammation and injury play an important role in the disruption of barrier function.[19,20] There is strong evidence for the role of cytoskeletal, focal adhesion and adherens junction proteins in regulating the EC barrier function.[28,29] In previous studies, others and we have demonstrated that exposure of ECs to ROS or edematic agents induce actin stress fibers, affect the focal adhesion and adherens junction proteins and alter the EC permeability.[17,22,24,27,30–32] Furthermore, exposure of lung ECs to ROS activates PLD resulting in the accumulation of PA and DAG which are derived from the catabolism of PC.[1,4,6–8,10,15,23] Our previous studies suggest that the activation of PLD either by oxidative stress or agonists such as thrombin or phorbol ester represents a key signaling pathway in the regulation of endothelial and epithelial function.[4,6,8,12,13,33]
In the current study, for the first time we demonstrated a role for PLD1 and PLD2 in the ROS-induced barrier function in lung ECs. Channeling the PLD-generated PA into PBt with 1-butanol and inhibition of PLD by C2-ceramide or blocking PLD1 and PLD2 with the catalytically inactive mutants (hPLD1-K 898 R and mPLD2-K758R), attenuated the H2O2-mediated ECs permeability, indicating a role for PLD activation in the agonist-mediated barrier dysfunction in ECs. Additionally, blocking PLD1 or PLD2 effectively attenuated the H2O2-induced formation of actin stress fibers and redistribution of VE-cadherin to the cell periphery. ROS such as H2O2 diperoxovanadate, superoxide and hydroxyl radicals have been established to injure ECs and regulate the EC paracellular permeability. Mechanisms of regulation of the ROS-induced EC barrier dysfunction have been described.[20,22,27,28,34,35] Earlier studies conducted by our laboratory and others have demonstrated that the ROS-induced permeability changes in the endothelium are dependent on signaling pathways involving the increase in intracellular calcium, activation of Rho, MLCK, PKC, Akt, MAPKs and Src kinases.[4,6–8,22,24,27,36–42] There is considerable evidence that many of these signaling pathways modulate the cytoskeleton, adherens junction assembly and cell-extracellular matrix attachments towards regulation of the EC barrier function.[2,19,20,28,29,35,38,43–46] Our current results on PLD1-and PLD2-dependent regulation of the ROS-mediated permeability involving actin stress fiber formation and VE-cadherin redistribution are in agreement with the involvement of cytoskeleton reorganization and tight junction proteins in LPA/S1P-induced endothelial permeability.[17] In this study, a selective role of PLD2, but not PLD1, in control of EC permeability by LPA or S1P involving the stress fiber formation and occludin down-regulation has been observed in HUVECs.[17] Similarly, a requirement of PLD activity for the actin stress fiber formation has been demonstrated in fibroblasts,[47] epithelial cells,[48] and aortic ECs.[49]
The mechanism(s) of PA-mediated EC barrier regulation is not completely defined. PA is a second messenger and mediates the biological and physiological functions of PLD activation either directly or indirectly by its conversion to LPA or DAG. In bovine lung ECs, ectopic PA has been shown to induce permeability changes that are dependent on the mobilization of intracellular calcium, FAK phosphorylation and tyrosine kinase activation.[40] However, it is unclear whether the ectopic PA-mediated permeability in ECs is mediated by a receptor-dependent or non-receptor pathway. Until now, PA-specific plasma membrane receptor(s) has not been identified in mammalian cells. PA generated intracellularly may modulate signaling cascades via non-receptor pathways. PA generated by PLD1/PLD2 pathway has been shown to activate PKC ζ 12 , alter actin cytoskeleton and modify actomyosin contraction.[49] Several studies have demonstrated an important role for RhoA family of GTPases, Rho, Rac and Cdc 42 in regulating the endothelial barrier function in response to agonists.[30,50–54] It is well recognized that actin polymerization leading to the formation of stress fibers is RhoA-dependent,[55] a process that is partly regulated by the PLD/PA signaling axis.[17,47,49,56–58] Interestingly, PLD2 has guanine nucleotide-exchange factor (GEF) activity for Rho and can regulate actin stress fibers in a manner independent of its lipase activity.[59–61] Additionally, PA activates phosphatidylinositol-4-phosphate 5 kinase (PI4P5K) to generate phosphatidylinositol-4,5-bisphosphate (PIP2),[62] an activator of actin cytoskeleton and modulator of interactions between actin and actin-binding proteins such as vinculin and filamin.[63] PLD-generated PA also activates Rac1 and IQGAP1, a process that is linked to the hyperoxia-induced NADPH oxidase activation and ROS generation.[23] Thus, the activation of PLD and generation of PA mediate the agonist- or ROS-induced endothelial permeability by modulation of Rho GTPases, PIP2 formation and actin cytoskeleton. As ROS activate PLD and PA generated by PLD signaling stimulates NADPH oxidase system that generates elevated levels of ROS, PLD and PA form both positive feedback and feedforward loops that amplify and integrate complex upstream and downstream signaling pathways. Further studies, at the systems biology level, are required to understand better how PLD/PA signaling axis regulates and coordinates the complex cellular functions in mammalian cells.
In summary, we have demonstrated a novel role for PLD1 and PLD2 activation and involvement in the ROS-mediated actin stress fiber formation and permeability changes in lung ECs (Fig. 6). These results provided the first direct evidence for the PLD mediation of ROS-induced lung injury that hinges on the lung vascular endothelial barrier dysfunction is clinically relevant in the onset and development of lung diseases during the exposure to high oxygen in critically ill patients requiring ventilation in the ICU.
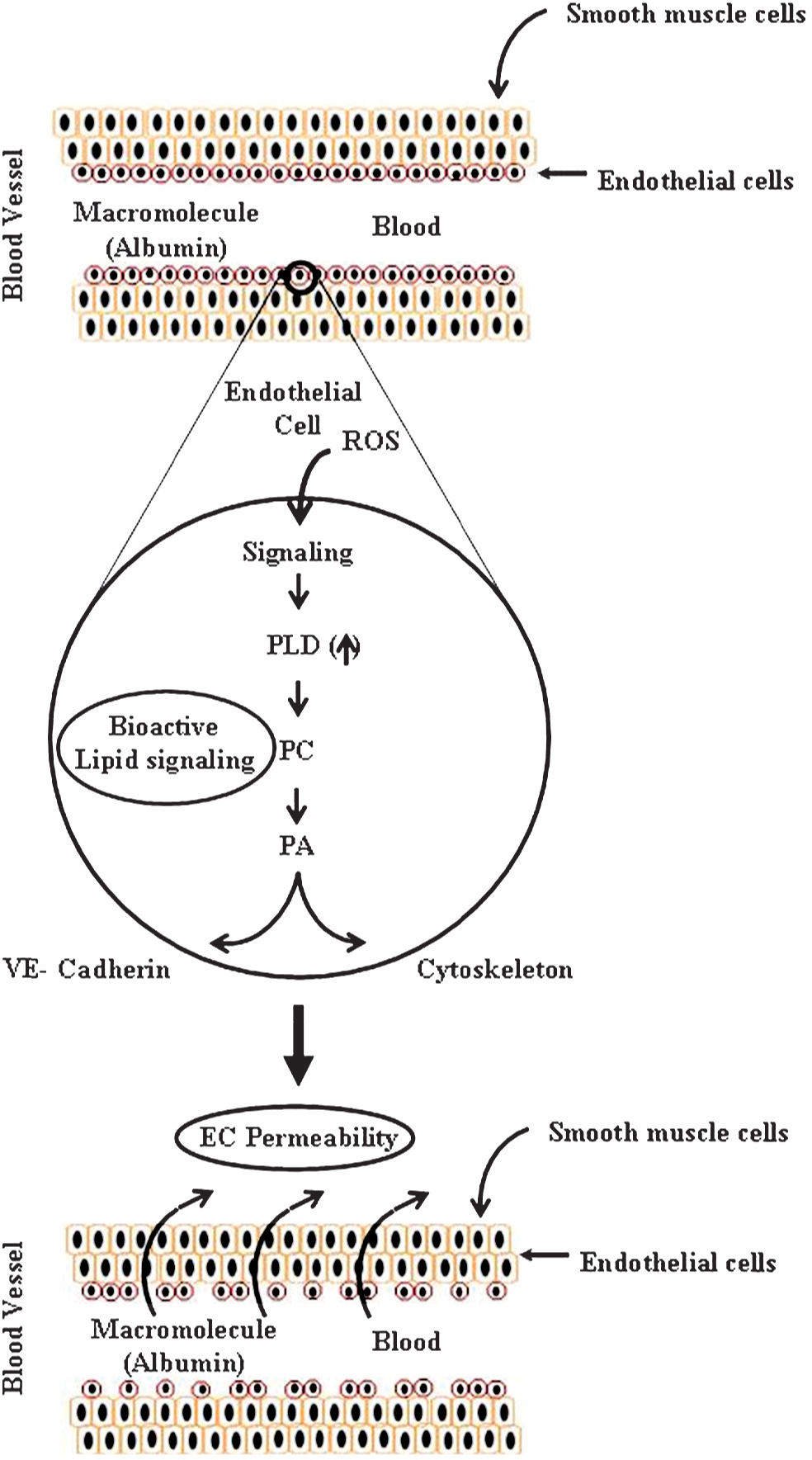
Phospholipase D signaling mediates reactive oxygen species-induced lung endothelial barrier dysfunction.