
Abstract
We previously reported an extremely rare case of follicular dendritic cell sarcoma (FDCS) presented as a thyroid mass. Given the rarity of this disease, there are no personalized and molecularly targeted treatment options due to the lack of knowledge in the genomic makeup of the tumor. A 44-year-old white woman was diagnosed with an extranodal FDCS in thyroid. The patient underwent a total thyroidectomy, central compartment dissection, parathyroid re-implantation, and adjuvant radiation therapy. Tumor DNA sequencing of 236 genes by FoundationOne panel found truncating mutations in PTEN and missense mutations in RET and TP53. However, patient-matched germline DNA was not sequenced which is critical for identification of true somatic mutations. Furthermore, the FoundationOne panel doesn't measure genomic rearrangements which have been shown to be abundant in sarcomas and are associated with sarcoma tumorigenesis and progression. In the current study, we carried out comprehensive genomic sequencing of the tumor, adjacent normal tissues, and patient-matched blood, in an effort to understand the genomic makeup of this rare extranodal FDCS and to identify potential therapeutic targets. Eighty-one somatic point mutations were identified in tumor but not in adjacent normal tissues or blood. A clonal truncating mutation in the CLTCL1 gene, which stabilizes the mitotic spindle, was likely a driver mutation of tumorigenesis and could explain the extensive copy number aberrations (CNAs) and genomic rearrangements in the tumor including a chr15/chr17 local chromothripsis resulted in 6 expressed fusion genes. The fusion gene HDGFRP3→SHC4 led to a 200-fold increase in the expression of oncogene SHC4 which is a potential target of the commercial drug Dasatinib. Missense mutations in ATM and splice-site mutation in VEGFR1 were also detected in addition to the TP53 missense mutation reported by FoundationOne.
Introduction
Originally described by Monda et al. in 1986, 1 follicular dendritic cell sarcoma (FDCS) originates from follicular dendritic cells at germinal center of lymphoid follicles that bind and retain immune complexities. 2 FDCS is an rare disease and accounts for less than 0.4% of all soft tissue sarcomas, 3 and around two thirds of FDCS 4 are localized within the lymph nodes. Extranodal sites of FDCS include the oral cavity, spleen, liver, bowel and thyroid. Extranodal thyroid FDCS is an exceedingly rare tumor with only three case reports to-date.5–7 There is a lack of genomic understanding of this disease resulting in the lack of personalized and molecularly targeted treatment options.
We previously reported the 3rd case of thyroid FDCS. 7 A 44-year-old white woman was diagnosed with an extranodal FDCS in thyroid. 7 The patient underwent a total thyroidectomy, central compartment dissection, parathyroid re-implantation, and adjuvant radiation therapy. Tumor DNA sequencing of 236 genes by FoundationOne panel found truncating mutations in PTEN and missense mutations in RET and TP53. However, patient-matched germline DNA was not sequenced which is critical for identification of true somatic mutations. 8 Furthermore, the FoundationOne panel doesn't measure genomic rearrangements which have been shown to be abundant in sarcomas and are associated with sarcoma tumorigenesis and progression. In the current study, we carried out comprehensive genomic sequencing of the tumor, adjacent normal tissues, and patient-matched blood, in an effort to understand the genomic makeup of this rare extranodal FDCS and to identify potential therapeutic targets.
In the current study, we performed comprehensive genomic profiling of this thyroid FDCS using the massive parallel Next Generation Sequencing (NGS) technology that include: i) exome sequencing of tumor, adjacent normal tissue, and patient-matched blood DNAs for the detection of small point mutations; ii) whole genome mate-pair sequencing of tumor, adjacent normal tissue, and blood DNAs to identify large genomic rearrangements; and iii) transcriptome sequencing of the tumor and adjacent normal tissue RNAs to quantify gene expression and identify expressed fusion genes. Our goal is to study the genomic makeup of this rare tumor and also hopefully provide insights into potential personalized treatment options.
Case Report
The exome sequencing identified 81 somatic mutations (Supplementary Table S1) in tumor but not in blood or adjacent normal tissues, giving a mutation rate of 1.2 per million base-pairs (Mbp) compared to the average rate of 0.41 per Mbp in papillary thyroid cancer. 9 Interestingly, out of the three mutated genes reported by FoundationOne: i) the RET mutation is a germline variant (Supplementary Figure 1) because it's clearly also present in the blood DNA as well as in tumor adjacent normal tissue; ii) two PTEN mutations that were supported by very low percentage of reads (1% and 3% respectively) in FoundationOne Panel were not observed in our data; iii) the TP53 mutation was detected by both FoundationOne and us. In addition, the FoundationOne Panel didn't report mutations in CLTCL1, VEGFR1, or ATM, which are of particular interests due to their established roles in cancer (Table 1, Supplementary Figure 2A-D).
Sequencing evidence of somatic point mutations in CLTCL1, VEGFR1, ATM and TP53.

The data is represented as numbers reads supporting mutation/total read depth (alternative allele fraction); or NA indicating no sequencing coverage. All mutations were observed in tumor DNA or RNA sequencing but not in germline or tumor adjacent normal tissue DNA/RNA sequencing.
The clonal heterozygous truncating mutation L433* in CLTCL1 was found in tumor exome sequencing and was consequently validated by Sanger sequencing (data not shown). While majority of the mutations detected in the tumor were subclonal [alternative allele concentrations (AAC)<0.4], the CLTC1 mutation with AAC of 0.63 is clearly clonal (Table 1; Supplementary Figure 3A) and happened early during tumorigenesis as a potentially driver events. CLTCL1 is involved in stabilization of the mitotic spindle 10 and the mutation L433* truncated the VPS functional domain leading to faulty assembly of clathrin complex in tumor (Supplementary Figure 3B-C).
The subclonal splice site mutation (AAC=0.22) in VEGFR1 detected in the exome located at an intronic splice-site 3bp from the exon boundary led to intron retention in expressed mRNA (Supplementary Figures 2D and 4) which consequently introduced an early stop codon in the kinase domain of VEGFR1. VEGFR1 could act as a decoy receptor to sequester VEGF away from VEGFR2, 11 hence a loss of function in VEGFR1 might lead to activation of the VEGFR2 pathway and drive angiogenesis.
In addition, subclonal missense mutations were identified in ATM (ACC=0.3; Supplementary Figure 2C) and TP53 (ACC=0.11; Supplementary Figure 2B) genes by both exome and RNA sequencing.
Extensive genomic structural changes were detected by whole genome mate-pair sequencing including CNAs and translocations/inversions (Supplementary Figure 5 and Supplementary Table S2). Two selected CNA regions with either moderate (chr8q) or high (chr17q) CN gains were experimentally validated using FISH (Supplementary Figure 5). Mostly interestingly, we observed a local chromothripsis event intra- and inter- chr15 and chr17 (Figure 1A), including multiple CNAs and rearrangements, resulted in 6 expressed fusion genes detected in RNA-Seq (Supplementary Table S3). Soft tissue sarcomas are known for high frequencies chromothripsis-like patterns in ∼80% of cases. 12 Of the 6 expressed fusion RNAs, two were of particular interest which we will discuss in detail below.
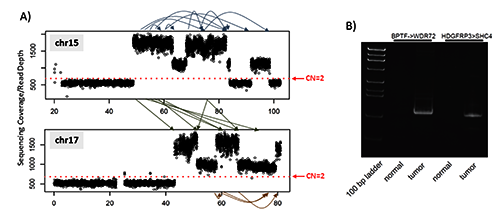
Local chromothripsis intra- and inter- chromosome 15 and 17 resulted in extensive copy number aberrations and expressed fusion transcripts. A) Whole genome mate-pair sequencing detected extensive genomic rearrangements intra- and inter- chr15 and chr17. The Y axis is the sequencing depth of the regions. The red dotted line indicates the sequencing depths representing normal copy number of 2 which is calculated across the whole genome from the mate-pair sequencing data. Arrows within and between chr15/chr17 showed the locations of rearrangements (detail in Supplementary Table S2); B) RT-PCR validation of the two expressed fusion transcripts. Both fusions were detected in tumor but not adjacent normal tissues.
Evidence for the fusion between HDGRFP3 and SHC4 was found in mate pair as well as RNA-Seq data (Supplementary Tables S2 and S3) and was confirmed with RT-PCR at the mRNA level (Figure 1B). The fusion gene is in-frame with the exon1 of HDGFRP3 fused in front of exon 2 of SHC4, and the putative fusion protein contains the first 28 amino acids of HDGFRP3 and SHC4 from amino acid 196 onwards (Figure 2). Expression of HDGFRP3 is much higher than SHC4 in tumor adjacent normal tissues based on the RNA-Seq data (Figure 2). However, the expression of SHC4 in tumor was over 200fold higher which is clearly due to fusion because only the fused-in exons had this drastic increase of expression (Figure 2). SHC4 is an oncogene and a member of the mammalian Src homology and collagen (SHC) family and works as the phosphotyrosine-binding docking molecule. Phosphorylation of SHC4 at the conserved tyrosine induces Ras and MAPK up-regulation.

Structural details of the fusion gene HDGRFP3->SHC4. The upper panel shows that the fusion gene consists of the 1st exon from HDGRFP3 and exons 2-12 from SHC4. The structure of the fusion protein is also illustrated. The lower panel shows the expression of HDGRFP3, SHC4, as well as the HDGRFP3->SHC4 gene in the RNA-Seq data of tumor and adjacent normal tissues.
A fusion between BPTF and WDR72 was found in both RNA-Seq and mate pair data (Supplementary Table S2 and S3) and confirmed by RT-PCR (Figure 1B). The first 18 exons of BPTF fused into the middle of exon 21 of WDR72 which introduced an early stop codon with the loss of the second PHD zinc finger and Bromo domains in BPTF (Figure 3). BPTF is part of the NURF complex which is associated with chromatin remodeling, and the second PHD zinc finger binds to H3K4me3, 13 associated with transcription start sites. The loss of this PHD zinc finger may result in transcriptional deregulation.

Structural details of the fusion gene BPTF->WDR72, where exons 1-18 from BPTF fused in front of the 2nd exon of WDR72.
Discussion and Conclusions
We presented the first extensive genomic characterization of an extranodal thyroid FDCS case using the genomic sequencing approach. This tumor was previously assayed by FoundationOne Panel which sequenced 236 cancer related genes in tumor tissue only without paired patient normal samples. 7 Our comprehensive genomic profiling of the tumor, tumor adjacent normal and patient germline DNA identified additional site mutations not reported by and large genomic rearrangements not targeted by the FoundationOne Panel. Specifically we detected mutations in CLTCL1, ATM and VEGFR1 in addition to the TP53 mutations reported by FoundationOne. Furthermore, the global approach allowed for detection of extensive copy number aberrations and genomic rearrangements including a chr15/chr17 local chromothripsis event with over 24 structural variants which resulted in 6 expressed fusion genes. The fusion gene of HDGFRP3→SHC4 led to the overexpression of a potential oncogene, SHC4, and the fusion between BPTF→WDR72 resulted in the truncation of BPTF functional domains involved in chromatin remodeling.
The most intriguing discovery from this study was the HDGFRP3→SHC4 fusion which resulted in a 200-fold overexpression of the oncogene SHC4, a member of the Src family. The Src inhibitor Dasatinib has been successful treating soft tissue sarcoma 14 and papillary and anaplastic thyroid cancer 15 and might be a potential drug treating this tumor. Furthermore, SHC4 is downstream of the EGFR signaling pathway and a previous study 16 found that seven out of eight nodal FDCS cases showed moderate to high levels of EGFR; hence the EGFR pathway might be involved in the pathogenesis of this disease.
Footnotes
Acknowledgements
We would like to acknowledge the expert help from Mayo Cytogenetics Core and its Director Patricia T. Greipp, as well as Bruce Eckloff from Mayo Sequencing Core and Chris Kolbert from the Mayo Gene Expression Core. Support for this work was provided by generous gifts from Everett and Jane Hauck to the Center for Individualized Medicine at Mayo Clinic Jacksonville Florida; and by Donna Foundation.