
Abstract
Intravascular Large B-cell lymphoma (IVLBCL) is an exceptionally rare form of non-Hodgkin lymphoma (NHL) distinguished by the preferential growth of neoplastic cells within blood vessel lumen. Challenging to detect and deemed disseminated at diagnosis, this condition is characterized by a highly aggressive, inconspicuous course with a high mortality rate. We describe the case of a 48 year-old African-American female presenting with a two month history of low-grade fevers and malaise. Laboratory data was notable for anemia, thrombocytopenia, elevated liver function tests, and hematuria. An extensive work-up for infectious, rheumatologic and malignant causes was negative. Her symptoms progressed and within two weeks, she was admitted for disseminated intravascular coagulation (DIC). Her course was complicated by diffuse pulmonary hemorrhage and ultimately, care was withdrawn. Autopsy identified widespread CD-20 positive intravascular large B-cell lymphoma with significant hepatosplenic involvement, characteristic of the Asian variant IVLBCL. This case uniquely highlights development of the Asian variant IVLBVL in a previously undescribed race. Identified by its intraluminal vascular growth pattern, IVLBCL generally spares lymphatic channels. Diagnosis and differentiation of this condition from other hematological malignancies via skin, visceral and bone marrow biopsy is imperative as anthracycline-containing chemotherapies may significantly improve clinical outcomes. This article outlines the common presentation, natural course, and treatment options of IVLBCL, along with the histopathology, immunohistochemistry, and chromosomal aberrations common to this condition.
Keywords
Case Report
In June of 2008, a 48 year-old African-American woman presented to her primary care physician describing a three week history of low-grade fevers and malaise. Antibiotics were administered, with no resolution of symptoms. She was subsequently admitted for further work-up. On admission, her laboratory data was notable for anemia and thrombocytopenia. Initial testing included a broncheoalveolar lavage (BAL) and lumbar puncture, which were non-diagnostic. A liver biopsy was notable for a mononuclear infiltrate consistent with chronic hepatitis. Laboratory data was notable for a normal Coombs test, autoimmune and vasculitic panel, and hepatitis panel. Computed tomography of the chest, abdomen and pelvis (CT-CAP) was unremarkable. She was discharged one month later with no formal diagnosis.
Within two weeks, she was re-admitted for progression of symptoms. Admission laboratory data was notable for severe anemia (Hgb 6.0 g/dL), leukopenia (2.5 g/dL); thrombocytopenia (10×109), and an elevated lactate dehydrogenase (LDH; 1900 U/L; reference range 122-222 U/L). Aspartate transaminase (AST; 77 U/L), alanine aminotransferase (ALT; 46 U/L), total bilirubin (8.8 mg/dL) and direct bilirubin (6.2 mg/dL) were all noted to be elevated. Haptoglobin was low. Prothrombin time (PT), activated partial thromboplastin time (APTT) and d-dimer levels were elevated. A peripheral blood smear showed schistocytes and polychromasia. Findings were deemed consistent with microangiopathic hemolytic anemia and disseminated intravascular coagulation (DIC). She was treated with plasma exchange and high dose steroid administration (methylprednisolone 125 mg/day) without effect. An extensive work-up for viral, bacterial and fungal pathogens was again, non-revealing. Magnetic resonance cholangiopancreatography (MRCP) of the liver showed hepatosplenomegaly with intra-organ hemosiderin deposition. Repeat CT-CAP defined diffuse hazy opacities in both lungs, hepatosplenomegaly and mild thickening of the walls of the cecum and ascending colon (Figure 1). On repeat CT of the chest three days later, there was interval increase in the bilateral nodular opacities, air bronchograms, subcutaneous anasarca and findings consistent with pulmonary edema (Figure 2). Bronchoalveolar lavage (BAL) identified diffuse pulmonary hemorrhages. On hospital-day 5, the patient rapidly developed acute respiratory distress syndrome (ARDS) and pulse-less electrical activity (PEA). Despite successful resuscitation, she remained unresponsive and care was ultimately withdrawn.
Chest computed tomography: Admission CT displaying diffuse hazy opacities in both lung fields with occasional nodular formation.
Chest computed tomography: Day 3 CT displaying interval increase in bilateral nodular opacities, air bronchograms and pulmonary edema.
Bone marrow biopsy results performed earlier that week were available following her demise. Samples showed evidence of hyercellular (80–90% cellular) with trilineage hematopoiesis and a subtle atypical mononuclear cell infiltrate composed of large cells with slightly irregular nuclear contours and multiple visible nucleoli. An autopsy identified diffuse alveolar hemorrhage, bilateral renal congestion with acute tubular necrosis (ATN), hepatomegaly and a single enlarged perisplenic lymph node. Histological examination of multiple organs confirmed diffuse intravascular large B-cell type lymphoma (IVLBCL).
Histologic Andimmunohistochemical Findings
Histological examination showed capillary wall congestion with large; atypical lymphocytes (Figure 3). On higher magnification, lymphocytes displayed vesicular nuclei, prominent nucleoli and frequent mitotic figures (Figure 4). Some cells showed coarse nuclear chromatin or irregular and indented nuclei (Figure 4). Staining with common leukocyte antigen and B cell marker CD-20 exhibited strong uptake by neoplastic cells (Figure 5).
Capillary wall congestion with large atypical lymphocytes and diffuse hemorrhage. Hematoxylin and eosin stain, intermediate magnification.
Lymphocytes displaying vesicular nuclei, prominent nucleoli and frequent mitotic figures. Coarse nuclear chromatin and indented nuclei noted as well. Hematoxylin and eosin stain, intermediate magnification.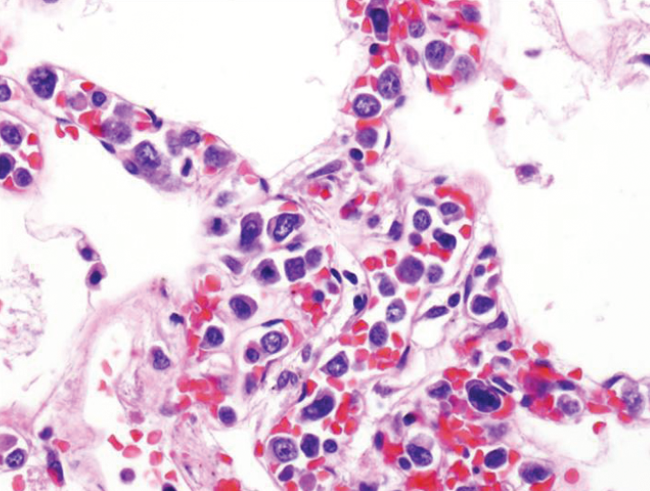
Tumor cell staining with B cell marker CD-20. High power magnification.
Discussion
First characterized by Pfleger and Tappeiner in 1959, 1 IVLBCL has emerged as an elusive malignancy characterized by a covert presentation and highly aggressive course. It is defined by the World Health Organization (WHO) as a rare and highly aggressive lymphoma variant, identified by the proliferation of large tumor cells within the lumen of small to medium-sized vessels. 2 We present the first reported case of the Asian variant IVLBCL in an African-American individual and proceed to review its unique presentation, progression and response to treatment.
Since its first description more than half-a-century ago, literature on IVLBCL has continued to accumulate. Until recently, the relative paucity of cases has limited epidemiologic research. However, it is currently recognized as a disease primarily of the elderly, with a median age in the sixth decade of life and a male-to-female ratio exceeding 3:1. 3 The wide variability in IVLBCL's clinical presentation makes detection challenging with the majority of patients present late within the disease process. Despite variability in presentation, specific patterns have been successfully traced to a surprising common entity: the geographic distribution of patients. Appropriately reflecting their origins, two main subtypes IVLBCL are recognized: the western variant and the Asian variant.
The western or classical variant remains the most common sub-type identified in North America and Europe. It is characterized by a prominence of neurologic and dermatologic findings, both at initial presentation and throughout its course. Neurologic symptoms may include alterations in consciousness and peripheral neuropathy. Cutaneous features may manifest as telangiectasias, diffuse ery-thematous streaks, purpuric macules and nodules. 4 The cutaneous variant of the western variant class is a unique subtype which is less aggressive than all other forms of IVLBCL, most commonly affecting pre-menopausal females.
The Asian or hemophagocytic variant, like it's counterpart, has a title reminiscent of its origin and is characterized by anemia (66%), thrombocytopenia (58%), hemophagocytosis (61%), bone marrow involvement (75%), respiratory symptoms (34%), B-symptoms (fever, night-sweats; 76%), hypoalbuminaemia (<30 g/L; 61%) and hepatosplenomegaly.5,6 In contrast to the Western variant, neurological symptoms (27%) and cutaneous lesions (15%) are rare and, when present, suggest an alternative diagnosis. 5 High lactate dehydrogenase (LDH; 98%) and disseminated intravascular coagulation (DIC; 25%) are common features.3,6 Most Asian variant IVLBCL cases have been reported in Southwestern Japan. A few case reports have described the hemophagocytic variant within the Western hemisphere. However, the majority of these have involved either immigrants from eastern countries or a known family history of Asian descent. Our case uniquely describes an African-American within the western hemisphere, presenting with the Asian variant.
Theories regarding the source of this the observed geographical difference remain disputed. More recent conjectures have included potential ethnic differences involved in inflammatory marker production such as sIL2R which appears markedly increased in Asian variant patients, in comparison to western variants. 3 Other theories speculate towards an environmental component given that the majority of Asian variant cases have been documented in Southwestern Japan where human T-cell lymphotropic virus type-1 (HTLV-1) is prominent. Helminthic infectious may also represent an environmental trigger. In contrast to other hemophagocytic-associated lymphomas, Epstein-Barr virus (EBV) has been reported in only a few IVLBCL cases and is not thought be causative.
The genetic alterations in IVLBCL remain under investigation. Aberrations in chromosomes 1, 6, 18, 11 and 13 have all been postulated as sources.3,7–11 However, in a retrospective analysis of cytogenetic abnormalities, Shimada et al. identified that only 57% of 84 patients displayed cytogenetic abnormalities. 3 Determination of the progenitor cell lines remains an area of intense investigation. Most cells express CD 19, CD20, and CD79a, consistent with B-cell origin. In addition to B cells, both intravascular T cell and natural killer (NK) cell lines have been identified and should not be considered a variant of IVLBCL. 12 It is plausible that Asian variant IVLBCL represents a unique variant of the germinal center B cell-like (GCB) form of CD5+ Diffuse Large B Cell Lymphoma (DLBCL).5,13 Accordingly, CD5– lines may be indicative of the western variant.
The paucity of extravascular migration remains IVLBCL's most perplexing feature. Defective interactions between luminal walls and lymphoma cells may play a role. 14 In healthy individuals, lymphocytes bind to high endothelial venules (HEV) on vessel walls via the lymphocyte homing receptor, thereby allowing transverse movement across the walls into peripheral regions. Western variant IVLBCL, in particular, appears to congregate in regions with low HEV, such as the brain and skin. 14 HEV deficiency within vessel lumens may thus prevent extravasation into the periphery. Other possibilities include deficiencies in essential transvascular migration molecules such as CD29 (β1-Integrin), CD54 (intracellular adhesion molecule 1), and CD18 (Integrin β2) superficially expressed by lymphoma cells.15,16 In the Asian variant, the mechanism of hemophagocytosis remains poorly understood, but may be influenced by macrophage colony stimulating factor and IL-6. 9 It is possible that hemophagocytosis is linked to cellular extravasation as studies have shown that the presence of hemophagocytosis correlates with disease dissemination (along with B-symptoms and elevated levels of soluble IL-2 receptor). 5 Notably, although the majority of IVLBC cases involve small vessels, at least seven case reports have identified IVLBCL in large vessels. 17 The mechanism of this is unknown.
As stated previously, IVLBCL may present with wide clinical variability. Characteristic physical exam findings associated with lymphoma, such as lymphadenopahthy or tumor burden, are often absent thus requiring greater reliance on laboratory data. Suspicion should be raised in patients describing headaches, skin abnormalities, hepatosplenomegaly, or constitutional complaints. Initial laboratory tests should include a complete blood count, serum LDH, β-2 microglobulin, monoclonal serum protein, a complete hepatic and renal panel and thyroid function studies. Cytogenetic studies should also be considered. The gold standard of diagnosis remains organ biopsy. Diffuse spread of the disease allows for physician discretion in determining the biopsy site. For Asian IVLBCL, bone marrow (BM) biopsy offers the most reliable results as intrasinusoidal tumor involvement is frequently encountered (up to 96%).6,9 Serial BM biopsies have been shown to enhance diagnostic accuracy. 18 Despite infrequent cutaneous features, Asian-variant IVLBCL is commonly isolated from skin biopsies and if positive, may prove a simple and useful diagnostic procedure. 6 Transbronchial biopsy may be performed if pulmonary symptoms are present. Hepatic and splenic biopsies are useful in the setting of hepatosplenomegaly and show sinusoidal involvement in up to 89% of patients. 9
Tumor cells are large with scant basophilic or amphophilic cytoplasm and moderately dispersed chromatin. Frequent mitotic figures and prominent vesicular nucleoli are common. Cells are rarely detected in peripheral blood. Sinusoidal distribution often occurs within the bone marrow, liver and spleen. As mentioned, erythrocyte-hemophagocytic cells are detected in the vast majority of BM biopsies. However, in the appropriate clinical context, the presence of classic findings including thrombocytopenia, fever and hepatosplenomegaly may be considered diagnostic even in the absence of hemophagocytosis. Computed tomography (CT) scans are rarely diagnostic. However, reports regarding the use of 18-Fluorodeoxyglucose-Positron Emission Tomography (FDG-PET) have been promising with characteristic uptake in the bone marrow and involved organs.19,20 The overall prognosis for most IVLBCL cases remains poor. Historically, most patients have been diagnosed with stage IV disease, or post-mortem. Post-mortum diagnosis appears less common in Asian variants (36%) versus the Western variants (64%). 5 Prognostic comparisons between the Western and Asian variants are similar, as are responses to treatment. 5 The rarity of IVLBCL has made randomized controlled trial assessment difficult. Historically, combination chemotherapy, typically CHOP, has been utilized with dismal results. However, the addition of Rituximab, a monoclonal antibody to CD-20, has led to substantial improvements in recovery, nearly doubling survival length. 21 European studies have determined an 81% survival rate at 3 years with combined immunochemotherapy, 22 versus just 33% in the absence of rituximab. 23 To date, no studies have evaluated the addition of Rituximab specifically in the Asian variant IVLBCL. Autologous stem cell support (ASCT) has been evaluated in a number of small studies and shown efficacy, primarily in individuals undergoing their first remission.3,23 However, the advanced age of most patients excludes them from this option. Allogenic peripheral blood stem cell transplant has been utilized with variable results in the setting of failed chemotherapy. 24 With treatment, Asian variant IVLBCL survival times range 12.5 months to 22.5 months.21,25 Cases exclusive to the skin appear to have a better response rate (3-year survival 56%). 26 Median survival with anthracycline therapy is estimated to be 13 months. 5 Etoposide has been effective in the setting of hemophagocytic syndrome. 27
Conclusions
Despite more than 50 years of research, IVLBCL remains a highly elusive malignancy typified by limited treatment options and a gaurded prognosis. We describe the first recorded occurrence of Asian variant IVLBCL in an African-American within the western hemisphere. This case is of particular interest as descriptions of the Asian variant IVLBCL in non-Asian populations is limited. No obvious mechanism has yet surfaced explaining this unique phenomenon. However, environmental factors characteristic to certain topographies may play a lesser role than once previously considered. Alternatively, African and Asian races may share particular genomic sequences predisposing them to the Asian variant. Regardless of the cause, it remains imperative that continued research be promoted to better understand this unique malignancy.