
Abstract
Adoptive cell transfer (ACT) of antigen (Ag)-specific CD8+ cytotoxic T lymphocytes (CTLs) is a highly promising treatment for a variety of diseases. Naive or central memory T-cell-derived effector CTLs are optimal populations for ACT-based immunotherapy because these cells have a high proliferative potential, are less prone to apoptosis than terminally differentiated cells, and have the higher ability to respond to homeostatic cytokines. However, such ACT with T-cell persistence is often not feasible due to difficulties in obtaining sufficient cells from patients. Here we present that in vitro differentiated HSCs of engineered PSCs can develop in vivo into tumor Ag-specific naive CTLs, which efficiently suppress melanoma growth. Mouse-induced PSCs (iPSCs) were retrovirally transduced with a construct encoding chicken ovalbumin (OVA)-specific T-cell receptors (TCRs) and survival-related proteins (i.e., BCL-xL and survivin). The gene-transduced iPSCs were cultured on the delta-like ligand 1-expressing OP9 (OP9-DL1) murine stromal cells in the presence of murine recombinant cytokines (rFlt3L and rIL-7) for a week. These iPSC-derived cells were then intravenously adoptively transferred into recipient mice, followed by intraperitoneal injection with an agonist α-Notch 2 antibody and cytokines (rFlt3L and rIL-7). Two weeks later, naive OVA-specific CD8+ T cells were observed in the mouse peripheral lymphatic system, which were responsive to OVA-specific stimulation. Moreover, the mice were resistant to the challenge of B16-OVA melanoma induction. These results indicate that genetically modified stem cells may be used for ACT-based immunotherapy or serve as potential vaccines.
Keywords
Introduction
Recent advances demonstrate that immunotherapy based on the adoptive cell transfer (ACT) of naturally occurring or gene-engineered T cells can mediate tumor regression in patients with metastatic cancers (59) or eliminate pathogens in patients with chronic viral infection (52,72). Current immunotherapies are based on the ACT of autologous tumor-infiltrating lymphocytes (TILs), administered in conjunction with interleukin-2 (IL-2) following host conditioning regimens (e.g., lymphodepletion). Although immunotherapies based on the ACT of autologous TILs are the best available treatment for patients with metastatic melanomas, the efficacy of naturally occurring TILs is mainly restricted to melanomas and does not extend to other tumors (3,69). Thus, current clinical trials utilize genetic modification of T cells for immunotherapy using a variety of techniques to express major histocompatibility complex (MHC)-restricted antigen (Ag)-specific T-cell receptor (TCR) or MHC-unrestricted chimeric Ag receptor (CAR) with a high affinity and specificity for target tumor-associated Ags (TAAs). Genetic engineering targets nearly all cancer histology because T cells can be engineered to target Ags expressed by transformed and nontransformed cells from within the tumor mass (56). Genetically engineered T cells can be used to combat all types of cancers or Ag-expressing cells; however, the biggest challenge for this approach is the identification of tumor-specific Ags that can be targeted without causing indirect toxicity to normal tissues (58).
Strong arguments support the development of cell-based therapies using genetically engineered T cells (12,21,31). While clinical trials show safety, feasibility, and potential therapeutic activity of cell-based therapies using this approach, there are concerns that undesirable effects arising from autoimmunity due to cross-reactivity from mispairing TCRs (35,70), off-target Ag recognition by non-tumor-specific TCRs (8), or on-target off-tumor toxicity by CARs (15,41) with healthy tissues could occur. In addition, genetically modified T cells using current approaches are usually intermediate or later effector T cells, which only have short-term persistence in vivo.
To date, stem cells are the only source available to generate a large number of naive Ag-specific T cells (22,39,44,75). Hematopoietic stem cells [HSCs; e.g., cluster of differentiation 34-positive (CD34+) stem cells] have a greater potential to pass the bone marrow barrier and travel in the blood; the use of HSCs for therapeutic purposes has been widely applied clinically, especially in HSC transplantation (42,48). However, HSCs have reduced differentiation and proliferative capacities, and HSCs are difficult to expand in cell culture (7,26). In fact, the ability to expand the number of HSCs in vivo or in vitro would represent a major advance to the most current and future medical uses of HSCs. In addition, HSC defects have been observed in the aged population and in disease conditions such as a bone marrow failure in Fanconi anemia patients (4,11,47). Pluripotent stem cells (PSCs) could serve as an effective alternative to the use of HSCs. Although the gold standard for PSCs remains embryonic stem cells (ESCs), their acquisition from patients is not feasible. In contrast, induced PSCs (iPSCs) can be easily generated from a patient's somatic cells by transduction of various transcription factors and exhibit characteristics nearly identical to those of ESCs (34,43,46). This approach provides an opportunity to generate patient- or disease-specific PSCs (14,55). Recently, a number of genetic methods as well as protein-based approaches have been developed to produce iPSCs with potentially reduced risks, including reduced immunogenicity (40,74). Our laboratory has published results indicating that the generation of Ag-specific T cells from iPSCs (i.e., iPSC-T cells) can be used for cell-based therapies of cancers and autoimmune disorders (22,36,37,39). In addition, we showed that overexpression of survival-related genes [e.g., B-cell chronic lymphocytic leukemia (CLL)/lymphoma-extra large (BCL-xL) and survivin] promotes T-cell persistence and tumor regression (38). Therefore, we hypothesize that genetically modified iPSCs with Ag-specific TCRs and survival-related genes, followed by differentiation driven by Notch signaling, will enable the iPSCs to pass hematopoietic and T-lineage differentiation checkpoints, resulting in the development of highly reactive Ag-specific CD8+ T cells.
Here we present that genetically engineered iPSCs with chicken ovalbumin (OVA)-specific TCRs and survival-related proteins demonstrated the ability to develop highly reactive OVA-specific cytotoxic T lymphocytes (CTLs), which were responsive to OVA-specific stimulation. Moreover, ACT-based immunotherapy in mice using such genetically engineered stem cells efficiently prevented the tumor growth of B16-OVA melanomas. These results indicate an alternative approach to develop highly reactive Ag-specific CTLs from genetically engineered stem cells with Ag-specific TCRs and survival-related proteins, which has a great potential to be used in ACT-based immunotherapy and vaccine discovery.
Materials and Methods
Ethics Statement
All animal experiments were approved by the Pennsylvania State University College of Medicine Animal Care Committee (IACUC protocol #42467) and were conducted in compliance with the guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
Cells and Mice
The male mouse iPS-MEF-Ng-20D-17 cell line was obtained from the RIKEN Cell Bank (Ibaraki, Japan) (39). The expression of octamer-binding transcription factor 3/4 (Oct3/4), sex-determining region Y box 2 (Sox2), Krüppel-like factor 4 (Klf4), and v-Myc avian myelocytomatosis viral oncogene homolog (c-Myc) was confirmed by RT-PCR as previously described (68), and green fluorescent protein (GFP) expression was confirmed by flow cytometry during the course of this study. The EL4 and OVA-expressing B16 melanoma (B16-OVA) cell lines (38,73) were obtained from Dr. Michael Croft (La Jolla Institute for Allergy and Immunology, La Jolla, CA, USA). OP9-DL1 cells [OP9 stromal cells expressing the Notch ligand delta-like 1 (DL1)] were obtained from Dr. Juan Carlos Zuniga-Pflucker (Department of Immunology, University of Toronto, Sunnybrook Health Sciences Center, Toronto, ON, Canada) (36). Both male and female OT-I TCR transgenic, female C57BL/6 (B6), and female Thy1.1 congenic mice (B6.PL-Thy1a/CyJ) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). In the experiments of ACT and tumor challenge, six female mice per group were used.
Peptides, Chemical, and Antibodies
OVA257–264 (SIINFEKL) and OVA323–339 (ISQAVHA AHAEINEAGR) peptides were purchased from the American Peptide Company (Sunnyvale, CA, USA). Mitomycin C (M0503) was purchased from Sigma-Aldrich (St. Louis, MO, USA). BCL-xL (#2762; recognizes both human and mouse) Ab was purchased from Cell Signaling Technology (1:1,000; Beverly, MA, USA). Phycoerythrin (PE), PE/cyanine 7 (Cy7), peridinin chlorophyll protein complex (PerCP), PerCP/Cy5.5, or allophycocyanin (APC) conjugated THY1.2 (1:1,000; clone 53-2.1), TCRVβ5 (1:1,000; MR9-4), TCRVα2 TCR (1:1,000; B20.1), CD25 (1:1,000; clone 3C7), CD69 (1:1,000; clone H1.2F3), CD62L (1:1,000; clone MEL-14), CD127 (1:1,000; clone A7R34), cytotoxic T lymphocyte-associated protein 4 (1:1,000; CTLA4; clone UC10-4B9), programmed cell death 1 (PD1; 1:1,000; clone 29F.1A12), CD44 (1:1,000; clone IM7), interferon (IFN)-γ (1:1,000; clone XMG1.2), IL-2 (1:1,000; JES6-5H4), carboxyfluorescein succinimidyl ester (CFSE; #423801), and propidium iodide (PI) (#421301) were purchased from Biolegend (San Diego, CA, USA). Annexin V:PE Apoptosis Detection Kit (559763) and Cytofix/Cytoperm™ (555028) were obtained from BD Biosciences (San Jose, CA, USA). Survivin (1:500; D-8, sc-17779; recognizes both human and mouse) and β-actin (1:800; C2, sc-8432) antibodies (Abs) for Western blotting were purchased from the Santa Cruz Biotech (Dallas, TX, USA). Mouse α-Notch2 Ab was kindly provided by Dr. Hideo Yagita (5 mg/ml; Juntendo University School of Medicine, Tokyo, Japan) (33,64).
Cell Cultures
The iPSCs were maintained on feeder layers of irradiated SNL76/7 cells (ATCC, Manassas, VA, USA) as previously described (36). OVA-specific iPSC-CTLs were plated in 48-well plates (Corning Costar, Corning, NY, USA), containing 1 ml Roswell Park Memorial Institute (RPMI)-1640 (Invitrogen, Carlsbad, CA, USA) with 10% fetal calf serum (Omega Scientific, Tarzana, CA, USA) at a density of 5 ′ 105/ml with 2 ′ 106/ml Ag-presenting cells (APCs) in the presence of 1 μM of OVA257–264 peptide. APCs were obtained from the spleens of syngeneic nontransgenic B6 mice (female C57BL/6L) by depleting T cells and treating with mitomycin C (100 μg/ml) for 30 min at 37°C.
Retroviral Transduction
MiDR, MIDR-OVA TCR-IRES-DsRed (MiDR-TCR), and MIDR-OVA TCR-2A-BCL-xL-2A-survivin-IRES-DsRed (MiDR-TCR-BCL-xL-survivin; human BCL-xL and survivin) were generated previously (39,73), and the exogenous human BCL-xL is larger than the endogenous mouse form, so they can be distinguished (23). Retroviral transduction was performed as previously described (57). The expression of DsRed was determined by flow cytometry gating on GFP+ cells. DsRed+ GFP+ cells were purified by cell sorting using a MoFlo high-performance cell sorter (Dako Cytomation, Fort Collins, CO, USA). Genomic DNA from DsRed+GFP+ cells was analyzed for TCRVβ5 gene expression by PCR. The forward primer is ACGTGTATTCCCATCTCTGGACAT, and the reverse primer is TGTTCATAATTGGCCCGAGAGCTG. PCR was performed in 50-μl reaction volume containing 100 ng DNA. All PCR components were used, according to the manufacturer's instructions (Taq DNA Polymerase; New England Biolabs, Ipswich, MA, USA), together with 1 mM of each primer. Annealing temperature of 68°C with 2 mM MgCl2 and 30 cycles were used.
Immunoblot
Cells lysates were extracted and used for Western blotting as previously described (57).
Cytokine Secretion, Cell Recovery, and Proliferation
Cytokines were measured by enzyme-linked immunosorbent assays (ELISAs; Biolegend); T-cell survival in vitro was determined by trypan blue (Sigma-Aldrich) exclusion assay; and proliferation was measured in triplicate cultures by incorporation of [3H]thymidine (1 μCi/well; ICN Pharmaceuticals, Laval, QC, Canada) during the last 12 h of culture (73).
In Vitro Cytotoxicity Assay
EL4 cells were incubated in 10 nM-10 μM CFSE PBS solution for 10–15 min at room temperature, and the CFSE-labeled EL4 cells were used as target cells (39,73). For peptide loading, target cells were incubated with specific OVA257–264 or control OVA323–339 peptide (5 μg/ml). Target cells were seeded into 96-well plates (10,000 cells/well). IPSC control, iPSC-CTLs, or CTL controls were added at different effector to target (E:T) cell ratios (1:5, 1:10, 1:20) in triplicate. For test of background, wells contained target cells only. The plates were incubated at 37°C for 12 h before flow cytometric analysis. Prior to analysis, PI (15 μg/ml) was added to distinguish live and dead cells. The percentage of specific lysis was calculated as follows: cytotoxicity (%): [100% × dead targets/(dead targets + live targets)] (experiment) – [100% × dead targets/(dead targets + live targets)] (background).
ACT and Tumor Challenge
Pre-iPSC-CTLs (3 × 106) in phosphate-buffered saline (PBS; Sigma-Aldrich) were intravenously (IV) injected into 4-week-old female Thy1.1 congenic mice, and in the following days, mice were intraperitoneally (IP) injected with 0.25 mg agonistic α-Notch2 Ab, 5 μg mouse recombinant IL-7 (rIL-7) and 10 μg mouse recombinant Fms-like tyrosine kinase 3 ligand (rFlt3L; PeproTech, NJ, USA), or a mouse IgG/PBS control (Jackson ImmunoResearch, West Grove, PA, USA) twice a week. After 2 weeks, the development of OVA-specific TCRVβ5+Thy1.2+CD8+ T cells in the lymph nodes and spleen was determined by flow cytometry. For tumor challenge, 2 weeks after adoptive transfer, mice were subcutaneously (SC) challenged on the flank with 4 ′ 106 B16-OVA tumor cells in 200 μl PBS or PBS without tumor cells as control. The numbers of T cells were calculated based on total cell numbers in the spleen and draining lymph nodes (inguinal, mesenteric, and para-aortic), together with the percentages of Thy1.2+CD8+TCRVβ5+ cells visualized by using flow cytometry (39). In some experiments, Thy1.1 mice were subsequently infected IP with 5 × 106 plaque-forming units (PFU) of recombinant vaccinia viruses expressing the gene for OVA (VV-OVA), provided by Dr. Shahram Salek-Ardakani (La Jolla Institute for Allergy and Immunology, San Diego, CA, USA) (62) and given 20 ng rIL-2 (PeproTech) IP after tumor inoculation twice per day for 3 days. The volume of the tumor (mm3) was measured using a caliper by a blinded investigator and calculated as follows: V = length × width2 × 0.52. Mice were sacrificed when the tumor size reached 20 mm in any direction.
Histology and Immunofluorescence
Routine hematoxylin and eosin (H&E) staining was performed at an interval of every five serial sections. For immunological staining, tissue sections were fixed with acetone (Sigma-Aldrich) and incubated with 3% bovine serum albumin (BSA; Sigma-Aldrich) to block nonspecific protein binding (39). Sections were stained with fluorescein isothiocyanate (FITC) anti-mouse TCR Vα2 (1:1,000; Biolegend).
Statistics
The unpaired t-test was used for statistical analysis between groups, and significance was set at 5%. All statistics were calculated using GraphPad Prism (San Diego, CA, USA).
Results
Generation of Highly Reactive Ag-Specific iPSC-CTLs
Previously, we have shown that iPSCs have the ability to differentiate into Ag-specific CTLs (39). In addition, we demonstrated that BCL-xL [a downstream molecule of nuclear factor κ-light-chain enhancer of activated B cells (NF-κB)] and survivin (an inhibitor of apoptosis, IAP family protein) are survival-related genes in T cells (57,65). Therefore, we sought to combine these two approaches to generate highly reactive Ag-specific iPSC-CTLs, which can produce a persistent Ag-specific immune response. We used retrovirus-mediated transduction and transduced mouse iPSCs with MHC-I restricted (H-2Kb) OVA257-264-specific TCRs, BCL-xL, and survivin, followed by coculture on the OP9-DL1 cells. We used the mouse stem cell virus-based retroviral vector MiDR (39) to generate the TCR and BCL-xL/survivin-transduced iPSCs (Fig. 1A). Upon gene transduction, we visualized the DsRed expression by fluorescence microscopy (Fig. 1B), and sorted DsRed+GFP+ cells (Fig. 1C). We confirmed the TCR gene integration by PCR (left) and survivin/BCL-xL expression (right) in the sorted cells by Western blotting (Fig. 1D). To determine whether the gene-transduced iPSCs were capable of undergoing the differentiation of Ag-specific CTLs, we first analyzed the iPSC morphological growth in the presence of Notch signaling. After 7 days of culture with OP9-DL1 cells, the iPSCs differentiated into mesoderm-like colonies and became lymphocyte-like cells on day 14, observed as nonadherent grape-like clusters. On day 28, lymphocyte-like cells spread fully on the plate (Fig. 1E). By calculating cell numbers of iPSC-derived lymphocyte-like cells, we found an approximate 1,000-fold increase in the cell number. We therefore hypothesized that the iPSCs that are transduced with the genes of Ag-specific TCRs and survivin/BCL-xL and stimulated with the Notch ligand are capable of differentiating into Ag-specific CTLs. We analyzed the cell surface markers of the iPSC-derived cells. On day 28 of in vitro coculture, the iPSC-derived cells expressed CD3 and TCR, two T-cell markers; the CD3+TCRβ5+ populations expressed CD8 but not CD4 (Fig. 2A). We also determined that the iPSC-derived cells sustained survivin/BCL-xL expression, even after long-term in vitro stimulation with the Notch ligand as detected by Western blotting (Fig. 2B). Collectively, our results suggest that iPSCs have the ability to differentiate into highly reactive Ag-specific CTLs using the approach of gene transduction of Ag-specific TCRs and survival-related genes into iPSCs, followed by in vitro stimulation with the Notch ligand.

In vitro generation of highly reactive antigen-specific induced pluripotent stem cell-derived cytotoxic lymphocytes (Ag-specific iPSC-CTLs). (A) Schematic representation of the retroviral construct MiDR-TCRVα2-2A-TCRVβ5-2A-BCL-xL-2A-survivin expressing ovalbumin (OVA)-specific T-cell receptor (TCR), B-cell chronic lymphocytic leukemia (CLL)/lymphoma-extra large (BCL-xL), and survivin. Ψ, packaging signal; 2A, picornavirus self-cleaving 2A sequence; LTR, long terminal repeats. (B) The gene-transduced induced pluripotent stem cells (iPSCs) (GFP+DsRed+) were visualized by fluorescence microscopy (scale bars: 50 μm). Nanog promoter-driven green fluorescent protein (GFP) serves as a marker of the iPSCs. (C) GFP+DsRed+ iPSCs were transduced with the retroviral construct and GFP+DsRed+ iPSCs (middle) were analyzed by flow cytometry and sorted by a high-speed cell sorter (right). (D) GFP+DsRed+ iPSCs were sorted, and genomic DNA was analyzed for the TCRVβ5 gene by PCR (left). The expression of BCL-xL, survivin, and β-actin was determined by Western blotting. Data are representative of three independent experiments. (E) Morphology of T-cell differentiation on days 0, 7, 14, and 28 after cocultures of iPSCs transduced with genes of the TCR, BCL-xL, and survivin on the delta-like ligand 1-expressing OP9 murine stromal cells (OP9-DL1 cells) in the presence of murine recombinant Fms-like tyrosine kinase 3 ligand (rFlt3L) and recombinant interleukin-7 (rIL-7) (scale bars: 20 μm). Data are representative of three independent experiments.

Highly reactive Ag-specific iPSC-CTLs stably express BCL-xL and survivin. The iPSCs transduced with the MiDR-TCRVα2-2A-TCRVβ5 (MiDR-TCR) or MiDR-TCRVα2-2A-TCRVβ5-2A-BCL-xL-2A-survivin (MiDR-TCR-BCL-xL-survivin) were cocultured with the OP9-DL1 cells in the presence of murine rFlt3L and rIL-7. On day 28 of in vitro coculture, the CD3+TCRβ5+CD8+ T cells were analyzed by flow cytometry and Western blotting. (A) Flow cytometric analysis of cluster of differentiation 3 (CD3) and TCRβ5, after gating on live iPSC-derived cells, and CD4 and CD8, after gating on CD3+TCRβ5+ populations. Data are representative of three independent experiments. (B) The expression of BCL-xL, survivin, and β-actin was determined by Western blotting. Data are representative of three independent experiments.
Functional Analysis of Highly Reactive Ag-Specific iPSC-CTLs
To determine the functional status of Ag-specific iPSC-CTLs, we measured the proliferation, cytokine production, survival, and cytotoxicity of these iPSC-CTLs following Ag stimulation. On day 28 of in vitro coculture, we isolated single-positive (SP) CD8+TCRβ5+ iPSC-CTLs and stimulated them with T-depleted splenocytes pulsed with OVA257–264 peptide and assessed the proliferation, cytokine production, and survival. OVA-specific iPSC-CTLs transduced with BCL-xL and survivin displayed significantly enhanced proliferation (Fig. 3A) and greater recovering numbers over time than those transduced with only TCR genes (Fig. 3B). However, both groups of OVA-specific iPSC-CTLs exhibited similar cytokine production (Fig. 3C). The percentage of early apoptotic cells [annexin V+ 7-aminoactinomycin D negative (7-AAD–)] in OVA-specific iPSC-CTLs transduced with BCL-xL and survivin were similar (10.7% vs. 10.9%), but the percentage of late apoptotic cells (annexin V+ 7-AAD+) were dramatically reduced (47.6% vs. 8.7%) compared to those in the cells transduced with only TCR genes (Fig. 3D). In addition, both groups of OVA-specific iPSC-CTLs displayed similar Ag-specific cytotoxicity (Fig. 3E). These results suggest that Ag-specific iPSC-CTLs are highly reactive and likely to induce a persistent Ag-specific immune response after adoptive transfer.

Functional analyses of highly reactive Ag-specific iPSC-CTLs. On day 28 of in vitro coculture, the single positive (SP) CD8+TCRβ5+ iPSC-CTLs were sorted and stimulated by T-depleted splenocytes pulsed with OVA257–264 peptide, and the proliferation, cytokine production, survival, and cytotoxicity were assessed. In some experiments, the sorted SP CD8+TCRβ5+ iPSC-CTLs were adoptively transferred into C57BL/6 mice that had been SC injected in the flank region with B16-OVA melanoma cells for a week and irradiated prior to the cell transfer. Mice were subsequently IV infected with recombinant vaccinia viruses expressing the gene for OVA (VV-OVA) and IP administered rIL-2 after the cell transfer. (A) Proliferation on day 1 to day 7 was measured by pulsing with tritiated thymidine for the last 20 h. Data are mean cpm ± SD from triplicate cultures and are representative of three experiments (*p < 0.05, **p < 0.01, Student's unpaired t-test). (B) Survival based on recovery of DsRed+Vβ5+ T cells over time. Cell numbers present on day 0 were assigned a value of 100%, and cell numbers surviving on day 4 to day 8 were used to calculate the recovery percentage. The data represent the mean ± SD percentage change from three separate experiments (*p < 0.05, Student's unpaired t-test). Data are representative of three independent experiments. (C) IL-2 and interferon (IFN)-γ production were measured by enzyme-linked immunosorbent assay (ELISA) at 40 h. Data are representative of three independent experiments. (D) Apoptosis of DsRed+CD8+ T cells on day 6 based on staining of annexin V and 7-aminoactinomycin D (7-AAD) and analyzed by flow cytometry. Data are representative of three independent experiments. (E) In vitro cytotoxicity assay. The iPSC-CTLs or control cells were added at different effector to target cell (E:T) ratios. Analysis was performed after a 12-h incubation period. Data are mean ± SD from three wells and representative of three experiments. (F) Tumor growth was monitored over time. Tumor volume was calculated as follows: V = length × width2 × 0.52. Data are mean tumor size ± SD from six individual mice and representative of three experiments. (G) Mouse survival was assessed over 25 days (Kaplan–Meier survival analysis).
To determine the functional status of Ag-specific iPSC-CTLs in a physiologically and clinically relevant setting, we used a murine model of melanomas. We SC injected C57BL/6 mice in the flank region with B16-OVA melanoma cells that express the OVA Ag recognizable by OVA257–264-specific CTLs. One week after tumor cells were injected, we irradiated mice and performed IV adoptive transfer of OVA-specific iPSC-CTLs into the recipient mice. In addition, we IV infected mice with recombinant VV-OVA, after the cell transfer and IP administered rIL-2 twice per day for 3 days (17). The transfer of iPSCs containing the control MiDR vector had no effect in suppressing tumor growth, correlating with their reduced survival, and all mice died in 2 weeks after the tumor challenge. Mice receiving OVA-specific iPSC-CTLs transduced with BCL-xL and survivin had smaller tumor sizes compared to those receiving the iPSC-CTLs transduced with only TCR genes or vector control (Fig. 3F), correlating with their enhanced survival (Fig. 3G). Collectively, these findings indicate that the ACT of highly reactive tumor Ag-specific iPSC-CTLs can suppress the growth of established tumors and operate therapeutically to control tumors.
In Vivo Development of Highly Reactive Ag-Specific iPSC-CTLs
Although mature Ag-specific iPSC-CTLs have promising therapeutic effects on ACT-based immunotherapy, their effective use in the clinic may be limited by complex and expensive in vitro differentiation approaches. In addition, the duration for generating iPSCs may limit their use for individualized treatment. Our previous study showed that TCR gene-transduced iPSCs developed into Ag-specific CTLs in vivo (39). We tested whether the OVA TCR and BCL-xL/survivin gene-transduced iPSCs have the ability to differentiate into highly reactive OVA-specific CTLs in vivo. We cocultured the OVA TCR and BCL-xL/survivin gene-transduced iPSCs (Thy 1.2+) on the OP9-DL1 cells for 7 days and then adoptively transferred the iPSC-derived cells into the recipient congenic mice (Thy 1.1+). We IP injected agonistic α-Notch2 Ab (33,64) and recombinant cytokines (e.g., rIL-7, rFlt3L) twice a week to boost the development of OVA-specific iPSC-CTLs in vivo. After 2 weeks, we analyzed Thy1.2+ TCRVβ5+ cells in the lymph nodes and spleen, gating on CD8+ T-cell population. The approach resulted in approximately 50% of OVA-specific iPSC-CTLs (Fig. 4A), which did not express CD25, CD69, CTLA4, and PD1, but expressed CD62L and CD127 (Fig. 4B), implying that the administration with Notch signaling promotes the development of naive Ag-specific iPSC-CTLs. Thy1.2+ TCRVβ5+ cells from the pooled lymph nodes and spleen were able to respond to Ag stimulation and produced IL-2 and IFN-γ (Fig. 4C). These results clearly demonstrate the in vivo development of highly reactive Ag-specific iPSC-CTLs using in vivo Notch signaling.

In vivo development of highly reactive Ag-specific iPSC-CTLs. The iPSCs transduced with the MiDR-TCR or MiDR-TCR-BCL-xL-survivin were cocultured with the OP9-DL1 cells in the presence of murine rFlt3L and rIL-7. On day 7, the DsRed+ cells that were sorted were adoptively transferred into Thy1.1 congenic mice, and on the following days, mice were IP injected with agonistic α-Notch2 Ab, rIL-7, and rFlt3L or a mouse IgG/PBS control twice a week. (A) After 2 weeks, Thy1.2+TCRVβ5+ cells from the pooled lymph nodes and spleen were analyzed by flow cytometry, after gating on CD8+ T-cell population. A representative image from mice receiving the iPSCs transduced with the MiDR-TCR-BCL-xL-survivin is shown following IgG control or agonistic α-Notch2 Ab, rIL-7, and rFlt3L protein injections. (B) Expression of CD25, CD69, CD62L, CD127, CTL4, and PD1 was analyzed by flow cytometry, after gating on CD8+Thy1.2+TCRVβ5+ T cells from the pooled lymph nodes and spleen (dark lines; shaded areas indicate isotype controls). Data are representative of three independent experiments. (C) IL-2 and IFN-γ production (dark lines; shaded areas indicate isotype controls). The pooled lymph nodes and spleen were stimulated with OVA257–264 peptide and analyzed by intracellular cytokine staining, after gating on Thy1.2+TCRVβ5+ cells. Data are representative of three independent experiments.
In Vivo Persistence of Highly Reactive Ag-Specific iPSC-CTLs
To determine the functionality of Ag-specific iPSC-CTLs generated by in vivo approach, we performed an in vitro cytotoxicity assay. OVA257–264-specific CTLs and CTLs from OT-I TCR transgenic mice displayed similar Ag-specific cytotoxicity and did not respond to nonspecific OVA323–339 stimulation (Fig. 5A). These results indicate that Ag-specific iPSC-CTLs generated by in vivo approach are functional.
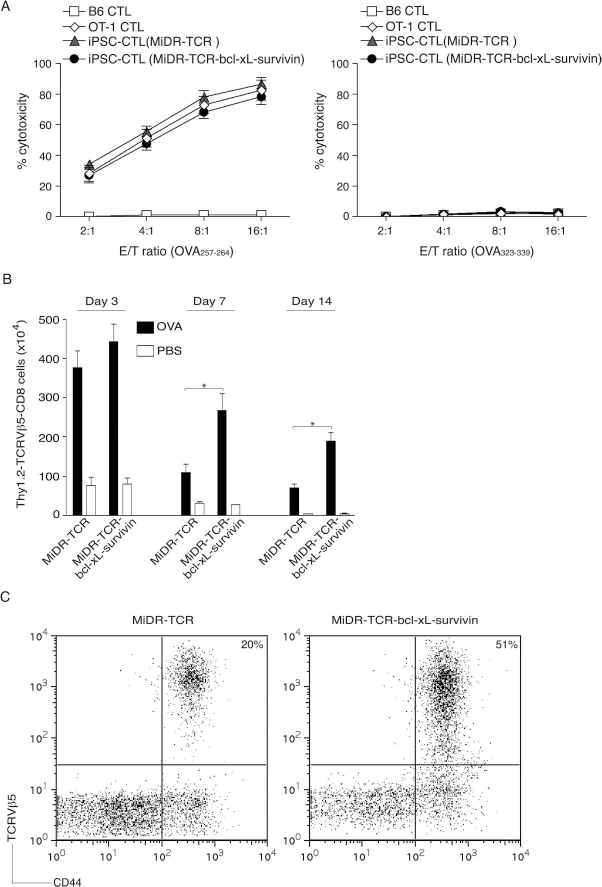
Highly reactive Ag-specific iPSC-CTLs persist in vivo. In vivo development of highly reactive Ag-specific iPSC-CTLs was performed as described in Figure 4. Two weeks later, after the iPSC transfer and the in vivo Notch signaling, CD8+Thy1.2+TCRVβ5+ T cells from the pooled lymph nodes and spleen were sorted, and an in vitro cytotoxicity assay was performed. In some experiments, mice were subsequently IP challenged with whole OVA protein (100 μg) in PBS (filled bars) or with PBS alone (open bars). (A) In vitro cytotoxicity assay. The iPSC-CTLs, OVA257–264-specific CTLs from OT-I TCR Tg mice, or nonspecific CTLs from B6 mice were added at different effector to target cell (E:T) ratios. Analysis was performed after a 12-h incubation period. Data are mean ± SD from three wells and representative of three experiments. (B) On days 3, 7, and 14, Thy1.2+TCRVβ5+CD8+ T cells were counted from the pooled lymph nodes and spleen. Data are the mean number of Thy1.2+TCRVβ5+CD8+ cells ± SD from six individual mice and representative of three experiments (*p < 0.05, Student's unpaired t-test). (C) At day 14, the percentage of TCRVβ5+CD44+ T cells was analyzed by flow cytometry, after gating on live CD8+ T cells in the spleen. Results are representative of three experiments.
To further determine whether Ag-specific iPSC-CTLs generated in vivo were capable of inducing the CTL persistence in response to Ag presented in vivo, we IP injected OVA protein into the mice that had received the pre-iPSC-CTLs and treated with agonistic α-Notch2 Ab and cytokines for 2 weeks. Mice receiving the OVA TCR and BCL-xL/survivin gene-transduced pre-iPSC-CTLs expanded more over 3 days in the lymph nodes and spleen than those pre-iPSC-CTLs expressing only TCR genes (Fig. 5B), suggesting the highly reactive Ag-specific iPSC-CTLs can readily proliferate and expand in vivo. The gene expression of BCL-xL and survivin enhanced numbers of Ag-specific T cells, whose peak response was on day 7 after Ag challenge. Additionally, these Ag-specific T cells were also present on day 14 post-Ag challenge after the secondary in vivo response was over and contraction of T-cell populations had occurred (Fig. 5B, C). Collectively, these results strongly support the conclusion that the in vivo development of transduced iPSCs leads to highly reactive Ag-specific iPSC-CTLs that have the ability to induce a persistent Ag-specific immune response.
Adoptive Transfer of Highly Reactive Ag-Specific Pre-iPSC-CTLs Suppresses Tumor Growth
To demonstrate that the highly reactive Ag-specific iPSC-CTL generated in vivo have the ability to induce Ag-specific CTL persistence in a physiologically and clinically relevant setting, we used a murine model of melanomas. We performed IV adoptive transfer of OVA-specific pre-iPSC-CTLs into the recipient congenic mice and IP injected agonistic α-Notch2 Ab as well as recombinant cytokines twice a week as described in Figure 4. Two weeks later, we SC injected in the flank region with B16-OVA melanoma cells. To generate better therapeutic effects, we also IV infected mice with VV-OVA after tumor inoculation and administered rIL-2 IP twice per day for 3 days (17). Three weeks after the tumor challenge, we observed more tumor-reactive CD8+ T cells infiltrating into melanoma tissues in mice receiving OVA-specific iPSC-CTLs transduced with BCL-xL and survivin than those receiving the iPSC-CTLs transduced with only TCR genes or vector control (Fig. 6). Similar to the therapeutic setting as described in Figure 3F, mice receiving OVA-specific iPSC-CTLs transduced with BCL-xL and survivin had the smaller tumor sizes compared to those receiving the iPSC-CTLs transduced with only TCR genes or vector control (Fig. 7A), correlating with their enhanced survival (Fig. 7B). Taken together, these findings indicate that the ACT of highly reactive tumor Ag-specific iPSC-CTLs can develop tumor Ag-specific CTL persistence and result in tumor protection.

Highly reactive Ag-specific iPSC-CTLs developed in vivo infiltrate into tumor tissues. In vivo development of highly reactive Ag-specific iPSC-CTLs was performed as described in Figure 4. Two weeks later, after the iPSC transfer and the in vivo Notch signaling, mice were SC injected in the flank region with B16-OVA melanoma cells that express the OVA Ag recognizable by the OVA257–264-specific CTLs. On day 21 to 22 after tumor challenge, tumor tissues were examined for tumor-reactive T-cell infiltration. (A) Hematoxylin and eosin (H&E) staining (scale bars: 20 μm). (B) Immunohistological staining (scale bars: 20 μm). OVA-specific TCRVα2+ CTLs (gray) infiltrated in OVA-expressing tumor tissues (the dark background). (C) Single-cell suspensions from tumor tissues were analyzed for expression of TCRVα2+ and TCRVβ5+ by flow cytometry, after gating on the CD8+ population. Data are representative of three independent experiments.
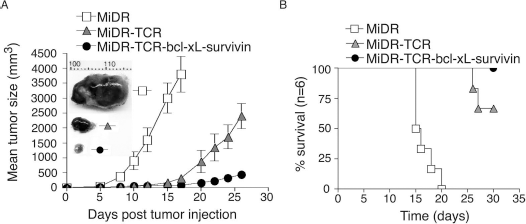
Highly reactive Ag-specific iPSC-CTLs developed in vivo suppress tumor growth. In vivo development of highly reactive Ag-specific iPSC-CTLs was performed as described in Figure 3. Two weeks later, after the iPSC transfer and the in vivo Notch signaling, mice were SC injected in the flank region with B16-OVA melanoma cells that express the OVA Ag recognizable by the OVA257–264-specific CTLs. Mice were subsequently IV infected with VV-OVA and IP administered rIL-2 after tumor inoculation twice per day for 3 days. (A) Tumor growth was monitored over time. Data are mean tumor size ± SD from six individual mice and representative of three experiments. Tumor representatives from the three groups at day 15 were shown. (B) Mouse survival was assessed over 30 days (Kaplan–Meier survival analysis).
Discussion
In this study, we demonstrated that genetically engineered iPSCs with Ag-specific TCR and survival-related proteins had the ability to differentiate into highly reactive and persistent Ag-specific CTLs. Moreover, we showed that ACT-based immunotherapy in mice using such genetically engineered stem cells efficiently prevented tumor growth in a murine model of melanomas. These results indicate that genetically modified stem cells have a therapeutic potential in adoptive immunotherapy.
The ACT of Ag-specific CTLs is a highly promising treatment for a variety of diseases. Ag-specific CTLs can secrete cytotoxins (e.g., perforin, granzymes) and cytokines (e.g., IFN-γ, TNF-α) or directly as well as indirectly kill/suppress the Ag-expressing cells. Naive or central memory T cell-derived effector CTLs are optimal populations for ACT-based immunotherapy because these cells have a high proliferative potential, are less prone to apoptosis than terminally differentiated cells, and have the highest ability to respond to homeostatic cytokines such as IL-7 and IL-15. However, such ACT with T-cell persistence is often not feasible due to difficulties in obtaining sufficient cells from patients. Here we present a new approach to generate highly reactive Ag-specific CTLs by genetically engineering iPSCs with Ag-specific TCR and survival-related proteins as well as stimulation with Notch signaling for ACT-based immunotherapy.
Intrinsic properties related to the differentiation state of adoptively transferred T-cell populations affect the success of adoptive immunotherapy. For ACT-based immunotherapy, the in vitro generation of naive or central memory T cell-derived effector cells for in vivo reinfusion is an optimal approach (16,27,28,32). However, current methodologies are limited in terms of the capacity to generate, isolate, and expand a sufficient quantity of such T cells from patients for therapeutic interventions.
Low numbers of Ag-specific T cells can be harvested from tumor masses and peripheral blood mononuclear cells (PBMCs), which contain T cells at different stages of differentiation: naive, early, intermediate, and late effector. Current expansion protocols with α-CD3-specific Ab plus rIL-2 or with specific Ag plus rIL-2 (the current clinical approach) drive differentiation of Ag-specific T cells in the intermediate and late effector stages and result in cells with shortened telomeres and diminished life span (60). In addition, many patients have decreased numbers or dysfunctional T cells in the peripheral blood or lesions. In this event, a patient's T cells cannot be used for in vitro expansion (1,54). In fact, T-cell survival in vivo is generally short-lived after they have been numerically expanded by in vitro culture.
TCR or CAR gene transfer has recently emerged as a method to overcome the obstacles of T-cell deletion and dysfunction in the clinic. Gene transduction of T cells from PBMCs with Ag-specific TCR (6,19,30,49) and CAR (2,20,50) elicits generation of functional CTLs and overcomes the challenge of the limited numbers of Ag-specific T cells. However, the engineered T cells express endogenous and exogenous polyclonal TCRs, which reduce their therapeutic potential (25,66). In addition, TCR mispairing is a concern with regard to the safety of TCR gene-transferred T cells for clinical use because the formation of new heterodimers of TCR can induce immunopathology [e.g., graft versus host disease (GVHD)] following ACT (5,51). Similarly, the engineered T cells expressing CAR can induce on-target off-tumor toxicity with healthy tissues (15,41).
For ACT-based immunotherapy, the “right” effector T cells from naive or central memory T cells are more attractive T-cell populations than intermediate and later effector T cells (9,18,27,28,61) because the differentiation state of T cells is inversely related to their capacity to proliferate and persist (10). The “right” effector T cells resist terminal differentiation, maintain high replicative potential [e.g., expression of common-γ chain (γc), CD132], are less prone to apoptosis (e.g., low expression of PD-1), and have the greater ability to respond to homeostatic cytokines, such as IL-7 and IL-15 (24,28,63,67,71), which facilitate their survival. In addition, such T cells express high levels of molecules that facilitate their homing to the lymph nodes, such as CD62L and chemokine C-C motif receptor 7 (CCR7). Furthermore, after providing an effective immune response, these effector T cells persist in a variety of differentiation states, providing protective immunity.
Last, harvesting sufficient numbers of Ag-specific, naive, or central memory T cells from patients' PBMCs for TCR or CAR gene transduction can be problematic due to the presence of very few cells. In addition, naive and central memory T cells are usually resistant to gene transduction unless they are activated. We have previously shown that BCL-xL (a downstream survival molecule of NF-κB) and survivin (an inhibitor of apoptosis) are survival-related genes in T cells (57,65). In this study, we employed the innovative convergence of genetic modification of PSCs with TCR/survival-related genes and stimulation with Notch signaling to produce large numbers of highly reactive Ag-specific PSC-T cells, which can target specific Ag. This study provides an improved tool to enhance the production and accelerate the use of stem cell-derived T cells as therapeutics.
There is the potential that the ACT of highly reactive Ag-specific iPSC-CTLs could trigger the development of leukemia or lymphoma because of the incorporation of oncogenes (i.e., BCL-xL and survivin). If this happens, a suicide gene, the inducible caspase 9 (iCasp9) (13,53), can be incorporated into the MiDR vector, as this allows the removal of the transferred iPSC-CTLs by the injection of a bioinert small-molecule dimerizing agent (AP1903) to “shut off” the system after tumor eradication. The tumor microenvironments can inhibit the T-cell development and function through the effect of suppressive cytokines, such as IL-10, or transforming growth factor (TGF)-β, inhibitory Tregs, or the negative costimulatory pathways involved in the tumor pathogenesis of T-cell exhaustion. Transferred Ag-specific iPSC-CTLs may become dysfunctional T cells, even overexpressing BCL-xL and survivin. If this occurs, neutralizing (α-IL-10 Ab), depleting (α-CD25 Ab), or blocking [(α-PD-1, α-CTLA-4, α-T cell immunoglobulin and mucin protein 3 (α-Tim-3), or α-lymphocyte-activation gene 3 (α-LAG-3) Ab] reagents can be used to manipulate the host microenvironment. In addition, CD4+ T cells help generate Ag-specific CTL memory. If this ACT-based immunotherapy does not result in persistent antitumor immunity, Ag-specific iPSC-CTLs can be adoptively transferred with CD4+ T cells generated from iPSCs. Alternatively, after the ACT, rIL-21 can be IP injected because rIL-21 can enrich for a population of central memory-type CTLs with “helper-independent” phenotype (29,45). Overall, the development of highly reactive tumor-specific iPSC-CTLs will be compatible with clinical application if the safety concerns using iPSCs are resolved.
In summary, we developed a new approach to generate stem cell-derived T cells that are likely to be naive and single-type Ag-specific T cells. In addition, we engineered the T cells with antiapoptotic genes that sustain T-cell survival/proliferation and to be highly reactive. As a result, the ACT with such stem cell-derived highly reactive T cells induces long-term T-cell persistence and results in immune surveillance.
Footnotes
Acknowledgments
This project was funded, in part, under grants with the National Institute of Health Grant R21AI109239 and K18CA151798, Leona M. and Harry B. Helmsley Charitable Trust 2014PG-T1D049, and the Pennsylvania Department of Health using Tobacco Settlement Fund SAP #4100057673 to J.S. The authors declare no conflicts of interest.