
Abstract
Severe neutropenia induced by chemotherapy or conditioning for hematopoietic cell transplantation often results in morbidity and mortality due to infection by opportunistic pathogens. A system has been developed to generate ex vivo-expanded mouse myeloid progenitor cells (mMPCs) that produce functional neutrophils in vivo upon transplantation in a pathogen challenge model. It has previously been demonstrated that transplantation of large numbers of freshly isolated myeloid progenitors from a single donor provides survival benefit in radiation-induced neutropenic mice. In the present work, an ex vivo-expanded and cryopreserved mMPC product generated from an allogeneic donor pool retains protective activity in vivo in a lethal fungal infection model. Infusion of the allogeneic pooled mMPC product is effective in preventing death from invasive Aspergillus fumigatus in neutropenic animals, and protection is dose dependent. Cell progeny from the mMPC product is detected in the bone marrow, spleen, blood, and liver by flow cytometry 1 week postinfusion but is no longer evident in most animals 4 weeks posttransplant. In this model, the ex vivo-generated pooled allogeneic mMPC product (i) expands and differentiates in vivo; (ii) is functional and prevents death from invasive fungal infection; and (iii) does not permanently engraft or cause allosensitization. These data suggest that an analogous ex vivo-expanded human myeloid progenitor cell product may be an effective off-the-shelf bridging therapy for the infectious complications that develop during hematopoietic recovery following hematopoietic cell transplantation or intensive chemotherapy.
Introduction
Neutropenia induced by intensive chemo- or radiation therapy in patients undergoing treatment for malignancy or hematopoietic cell transplantation (HCT) is one of the most important risk factors for the development of life-threatening opportunistic bacterial or fungal infections (14,37). Neutropenia is also the leading factor limiting the use of cancer chemotherapy, resulting in significant declines in survival (18,19). Infectious complications from prolonged neutropenia increase the duration of hospital stay, as well as morbidity and mortality. Current therapies include antimicrobials, cytokine treatment, and granulocyte transfusions. None of these provide comprehensive protection, illustrating the need for novel approaches.
Progenitor cells restricted to the myeloid lineages are capable of partially restoring functional myeloid hematopoiesis. The restoration is transient since progenitors do not possess extended self-renewal ability (2,23). Myeloid progenitor populations purified from the bone marrow have been shown to protect neutropenic mice from lethal fungal and bacterial infections in syngeneic and allogeneic mouse models of HCT (1-3). Such myeloid progenitors freshly isolated from the bone marrow are also capable of inducing tolerance (13). It was subsequently shown that myeloid progenitors generated ex vivo were capable of providing myeloid cell support during severe neutropenia following lethal doses of radiation and improving overall survival (32).
The scarcity of myeloid progenitors in either human bone marrow or mobilized peripheral blood makes direct isolation of cells as a therapeutic product for clinical use impractical. To address this issue, we have developed a method to expand and differentiate hematopoietic stem cells (HSCs) ex vivo into a population of myeloid progenitors. As demonstrated herein these culture-derived mouse myeloid progenitor cells (mMPCs) can be cryo-preserved and administered upon thawing without compromising function.
We demonstrate that pooled, cryopreserved mMPCs from several MHC disparate mouse strains protect neutropenic mice from fungal challenge and increase survival in a cell dose-dependent manner. In vivo characterization of the expansion and differentiation potential of the mMPC product in allogeneic recipients shows that all mMPC donors contribute to transient hematopoiesis and give rise to a large number of differentiated progeny mostly restricted to myeloerythroid lineages. These results suggest that an analogous pooled human myeloid progenitor cell product derived from CD34+ cells may be an effective bridging therapy during the period of severe neutropenia following stem cell transplantation conditioning regimens or high-dose chemotherapy to prevent morbidity and mortality from opportunistic infections.
Materials and Methods
Mouse Strains
All mice were maintained under Institutional Animal Care and Use Committee-approved protocols and procedures. C57BL6/Ka and C57BL6/Ka-EGFP mice were bred in house. BALB/c and AKR were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and FVB from Charles River Laboratories (Wilmington, MA, USA).
BALB/c mice, 8-12 weeks old, were irradiated with 8.9-Gy radiation using a Faxitron CP-160 (Faxitron, Tucson, AZ, USA), delivering 0.71 Gy/min. Radiation was delivered in a split dose 3—4 h apart. In some experiments (Figs. 3 and 4A), mice were irradiated with 9.2 Gy at 1.71 Gy/min using a Mark I Cesium irradiator (J. L. Shepherd and Associates, San Fernando, CA, USA), also given in two doses at least 3 h apart. Antibiotics (106 U/ L polymixin B sulfate and 1.1 g/L neomycin sulfate; Sigma-Aldrich Fluka, St. Louis, MO, USA) were added to the water following irradiation.
Isolation of Hematopoietic Stem Cells
HSCs for mMPC generation were isolated as c-Kit+Thy-1.1lowSca-1+Lin-/low (22). HSCs from BALB/c mice for transplantation were isolated as c-Kit+Sca-1+Lin- as described previously (33). Flow cytometric sorting and analysis was performed using a three-laser FACSAria (BD Biosciences, San Jose, CA, USA). Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA).
Derivation of mMPCs
Sorted C57BL6/Ka, C57BL6/Ka EGFP, AKR, or FVB HSCs were cultured in a 24-well plate at 10,000 cells/ well in 500 μl X-VIVO 15 media (Lonza, Walkersville, MD, USA) supplemented with 50 ng/ml rmSCF, 5 ng/ml rmTPO (R&D Systems, Minneapolis, MN, USA), 30 ng/ml rmFlt3L (Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA), Primocin (InvivoGen, San Diego, CA, USA), β-mercaptoethanol (Sigma-Aldrich), and glutamax (Invitrogen). Some cultures were also given 10 ng/ml IL-6 (Biosource/Thermo Fisher Scientific, Waltham, MA, USA) as noted in corresponding figure legends. Two days after plating, 500 μl of fresh complete media was added. On day 4, the cells were transferred to wells of a six-well plate containing 500 μl of medium. On day six, 500 μl of media was removed and 500 μl/well fresh media added. On day 7, the mMPCs were divided between two wells of a six-well plate containing 500 μl media. On day 9, the C57BL6/Ka or C57BL6/Ka-EGFP mMPCs were harvested and cryopreserved. The wells containing AKR or FVB mMPCs were passaged as on day 7. AKR and FVB cells were harvested and cryopreserved on day 10. In some of the earlier cultures with C57BL6/Ka mMPCs, the cells were harvested after 7 days of culture. However, for later experiments, the slightly longer culture times were used that result in increased expansion. mMPCs were cryopreserved in X-VIVO 15 media containing 10% DMSO (Sigma-Aldrich), 25% FCS (HyClone/Thermo Fisher Scientific, Waltham, MA, USA) at 20 million cells/ml.
Clonogenic Assays
Clonogenic assays were performed in methylcellulose (Stem Cell Technologies, Vancouver, BC, Canada) as recommended by the manufacturer. Growth factors added were 50 ng/ml SCF, 10 ng/ml IL-6, 10 ng/ml IL-3 (Invitrogen), and 3 U/ml erythropoietin (Janssen Biotech, Horsham, PA, USA) in X-VIVO 15 media.
Transplantation
Frozen cells were thawed in a 37°C water bath. Post-thaw viability and cell counts were determined by trypan blue (Gibco/Thermo Fisher Scientific, Waltham, MA, USA) exclusion. mMPCs were suspended at the desired concentration and pooled at equal ratios for transplantation into anesthetized mice by retro-orbital intravenous injection.
For spleen colony-forming unit assays (CFU-S), C57BL6/Ka mMPCs were infused into irradiated BALB/c or C57BL6/Ka mice. Spleens were fixed by immersing in Tellyesniczky's fixative (42% ethanol, 6% formaldehyde, 3% glacial acetic acid) to count the nodules.
Preparation of Aspergillus fumigatus Conidia
A suspension of a clinical isolate of Aspergillus fumigatus (2) was prepared as previously described. Briefly, fungus was plated on Sabaroud's dextrose agar (BD Biosciences, Cockeysville, MD, USA) for 48 to 72 h at 37°C. The conidia were harvested by gentle scraping in 10 ml PBS (Gibco) with 0.05% Tween 80 (J.T. Baker/ Avantor, Center Valley, PA, USA). The resulting suspension was filtered to remove hyphae and was maintained at 4°C. The concentrations of live conidia were reestablished before each experiment by counting and limited dilution plating on Sabaroud's dextrose agar plates. Mice were infected with 120 Aspergillus conidia via tail vein injection of 150 μl of conidia solution. At the time of inoculation, 150 μl of the injection solution was plated to confirm the numbers of live conidia infused.
Flow Cytometric Analysis of Blood and Tissues
Blood was analyzed for chimerism using antibodies to the FITC-conjugated H-2Kb (50x dilution), H-2Kk (50x dilution), or H-2Kq (100x dilution) and biotin-conjugated H-2Dd (100x dilution) loci (BD Biosciences), and the lineage markers PE-Cy7-conjugated B220 (400x dilution), APC-conjugated CD3 (100x dilution), and Pacific Blue-conjugated CD11b (400x dilution) and Gr-1 (1,600x dilution) (eBioscience, San Diego, CA, USA). Appropriate controls were used to set gate parameters. Mice were perfused by injection into the left ventricle with HBSS (Gibco) with 10 mM EDTA (J.T. Baker) to collect the blood. The bone marrow plug was disas-sociated by drawing through a 25-gauge needle. The spleen was disassociated by homogenizing between glass slides and treatment with collagenase IV (Worthington, Lakewood, NJ, USA) for 30 min at 37°C. The liver was mechanically disassociated, and dead cells and hepato-cytes were removed by centrifugation through a 35% Percoll solution (Sigma-Aldrich). mMPC chimerism in the tissues was determined using antibodies to the FITC-conjugated H-2kb, H-2kk, H-2kq, and biotin-conjugated H-2Dd (BD Biosciences), and the lineage markers PE-Cy7-conjugated B220, APC-conjugated CD3, Pacific Blue-conjugated, or PE-Cy7-conjugated CD11b (1,600x dilution), Pacific Blue-conjugated Gr-1 (1,600x dilution), PE-conjugated Ter119 (800x dilution), PE-conjugated CD11c (200x dilution), APC-conjugated MHC II (1,600x dilution) (eBioscience), and PE-conjugated CD71 (50x dilution) (Biolegend, San Diego, CA, USA). To estimate total bone marrow cells, the four long bones of the hind legs were processed, and the cell count was multiplied by four, since the processed bones make up only 20-25% of total bone marrow (9).
To analyze donor-derived platelets, blood was collected in 300 μl PBS with 10 mM EDTA and diluted in PBS with 2% dextran sulfate (Pharmacia/GE Healthcare, Uppsala, Sweden) and centrifuged. Supernatant was collected and centrifuged again. The pellet was stained with biotin-conjugated CD61 (100x dilution) (eBioscience). The platelets were additionally stained with PE-conjugated CD41 (200x dilution), Pacific Blue-conjugated Gr-1 (1,600x dilution), and APC-conjugated streptavidin (100x dilution) (eBioscience).
Micrographs
Photographs of May-Grünwald/Giemsa (Sigma-Aldrich) stained cells were taken using a BX40 microscope and Q-Fire digital camera (Olympus, Center Valley, PA, USA). The images were acquired in and processed using Photoshop Elements 2.0 (Adobe Systems, San Jose, CA, USA). Postacquisition processing was limited to global use of the levels' command to optimize contrast and cropping to assemble the figure from several images.
Statistical Analysis
Statistical significance of survival curves was assessed using Kaplan-Meyer survival analysis, with differences between groups analyzed by the log-rank test (Graphpad Prism 4.0, La Jolla, CA, USA). Statistical significance was evaluated using unpaired t-test, with p < 0.05 considered to be statistically significant.
Results
Ex Vivo-Generated Myeloid Progenitors Can Be Cryopreserved and Retain Phenotypic Characteristics and In Vitro Function Postthaw
To develop a therapy that provides temporary but effective recovery of cells of the myeloid lineage with little to no long-term engraftment potential, we established culture conditions to produce large numbers of mMPCs, myeloerythroid-restricted progenitors containing very few mature myeloid cells, and few cells with multipotent potential (the ability to generate all hematopoietic lineages), from mouse HSCs. HSCs were purified by flow cytometric sorting and cultured in X-VIVO 15 medium supplemented with the cytokines SCF, TPO, and Flt-3L for 9 days (C57BL6/Ka) or 10 days (AKR and FVB). At the end of the culture period, cells were analyzed for expression of the markers c-Kit (CD117) and Sca-1, indicators of hematopoietic progenitors, as well as markers of myeloid commitment, CD11b and Gr-1, and lymphoid commitment CD3 (T cell) and B220 (B cell) (Fig. 1A). By morphology, the majority of cells showed signs of myeloid commitment while retaining the blast cell characteristics of stem and progenitor cells (Fig. 1B). Routine expansion of total cells was 842 ± 250-fold, whereas the expansion of progenitors (Lineage-/lowc-Kit+) was 427 ± 127-fold (n = 31). The composition of the culture typically consisted of 52 ± 10% Lineage-/lowc-Kit+ progenitor cells, with 6.5 ± 2.7% Lineage-/lowc-Kit+Sca-1+ phenotypic HSC and multipotent progenitors (n = 31). Approximately 79 ± 9% (n = 16) of the cultured cell population expressed low to high levels of CD11b, and a smaller portion, 26 ± 8%, coexpressed the granulocyte marker Gr-1, illustrating myeloid commitment of the cultures, though the cells remain morphologically immature. No evidence of lymphoid differentiation was observed (Fig. 1A).

Characterization and cryopreservation of culture-derived mMPCs. (A) Analysis of a culture-derived mMPCs from C57BL6/Ka HSC. At the end of the 9-day culture period, Lineage-/low cells (upper left) were further analyzed for expression of c-Kit and Sca-1 (upper right). Analysis for markers of myeloid (CD11b and Gr-1) and lymphoid (B220 and CD3) differentiation is shown on the lower panels. Shown are representative flow cytometry plots of live cells and numbers indicate percentage of live cells. (B) May-Grünwald/ Giemsa-stained cytospins of a day 7 culture. Most cells are immature (black arrows), though many are myeloid committed (white arrows). Low numbers of relatively mature megakaryocytes are present (M). (C) Percentage of Lin-c-Kit+ cells in day 9 (C57BL6/Ka) or 10 (AKR and FVB) mMPC cultures prefreeze and postthaw. Left: Example of plots and gating. Right: combined data (mean ± SD) from 31 experiments. There is no significant difference in percentage of Lin-c-Kit+ cells prefreeze and postthaw. (D) Plating efficiency in methylcellulose medium of fresh and cryopreserved mMPCs. The cells were sorted on a FACSAria as c-Kit-positive or negative cells and plated at 200 or 300 cells/ml. None of the prefreeze/postthaw comparisons reach statistical significance. Combined data from four independent experiments using cultures derived from AKR, C57BL6/Ka, and FVB (two experiments).
Unlike mature granulocytes, myeloid progenitors and committed precursors can be cryopreserved (4,5). To demonstrate that cryopreserved mMPCs retained their functionality, mMPCs were generated from HSCs and analyzed by flow cytometry prior to freezing. The cells were then cryopreserved and stored in the vapor phase of liquid nitrogen for at least 7 days. Sample vials were removed, rapidly thawed in a 37°C water bath, and washed twice with medium. Viable cell recovery typically exceeded 50%. Analysis by flow cytometry shows that the combined progenitor and HSC content was not significantly affected by the freeze/thaw cycle with 49 ± 12% Lineage-/lowc-Kit+ progenitor cells remaining postthaw (n=31) (Fig. 1C). Freeze-thaw also had no significant impact on the response in clonogenic colony formation assays in vitro (Fig. 1D).
mMPCs have been generated by culture durations between 5 and 11 days, with increased culture duration gradually shifting the culture composition to more mature progenitors (data not shown). To test whether or not multipotent progenitor activity is present in the cultures, CD45-congenic C57BL6/Ka mice were reconstituted with mMPC derived by 5- or 8-day cultures (Fig. 2). Control mice reconstituted with myeloid progenitors directly purified from the bone marrow (BM MPs) showed very limited donor cell engraftment at week 4 (progeny of these cells peak at 1 to 2 weeks postinjection). Reconstitution with cells that have been in mMPC cultures for 5 days (day 5 mMPCs) resulted in robust donor cell engraftment with continued production of myeloid and lymphoid donor cells 4 weeks posttransplantation. Because the progeny of bone marrow myeloid progenitors have been exhausted by this time point (BM MPs), the presence of short-lived myeloid donor cells at 4 weeks indicates that the transplanted day 5 mMPCs contained functional multipotent progenitors, which can generate myeloid progeny for more than 4 weeks. Cells that had been cultured for 8 days (day 8 mMPCs) showed very limited donor cell engraftment at week 4 even at three times the cell dose of day 5 mMPCs. The engraftment pattern was comparable to what was observed when using freshly isolated bone marrow myeloid progenitors (BM MPs). The decreasing level of short-lived myeloid donor cells seen at 4 weeks suggests that mMPCs contain decreasing numbers of functional multipotent progenitors with increasing culture duration.
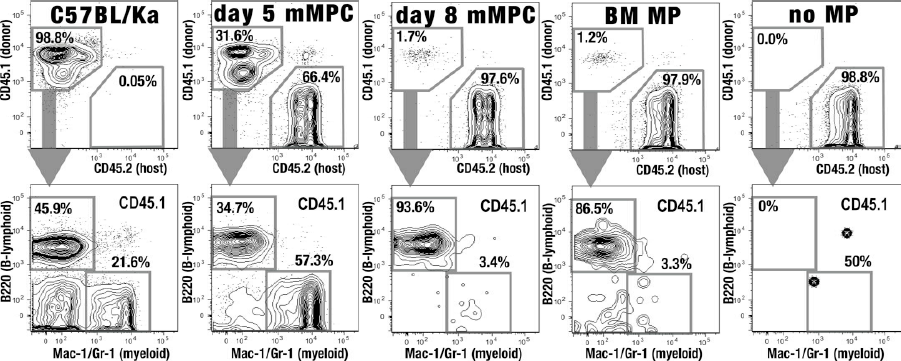
Engraftment of syngeneic mMPCs. Shown are the engraftment levels in peripheral blood at 4 weeks postreconstitution with 2.25 × 105 C57BL6/Ka cells cultured under mMPC conditions for 5 days (day 5 mMPC), 6 × 105 C57BL6/Ka cells cultured 8 days (day 8 mMPCs), or 2 × 104 myeloid progenitors sorted directly from mouse bone marrow (BM MPs). The engraftment obtained when injecting no cells (no MPs) is shown as a negative control, and the lineage distribution in donor animals (C57BL6/Ka) is shown as a positive control. Shown are the levels of CD45.2+ host cells and CD45.1+ donor cells in the blood indicating percentage of live cells (top) and the corresponding CD45.1+ donor cell distribution into B220+ B lymphoid and Mac-1(CD11b)/Gr-1+ myeloid cells (bottom).

mMPCs can rapidly protect neutropenic mice from a lethal fungal challenge. (A) Lethally irradiated BALB/c mice received 200 syngeneic HSCs only (black) or 200 syngeneic HSCs and 80,000 freshly isolated allogeneic myeloid progenitors (MPs) (thin gray) or 500,000 ex vivo-derived allogeneic mMPCs freshly isolated from the culture (bold gray) on the same day as irradiation. Fungus conidia were injected intravenously on day 7. The p values (log-rank comparison) compare the survival of each group with that in the HSC-only group. The p value in black compares survival between the myeloid progenitors (MP) and mMPC group. Myeloid progenitors (MPs and mMPCs) were used without prior cryopreservation. Radiation controls (dotted) and HSC rescue (dashed) were not exposed to fungus spores. Combined data from three experiments are shown. (B) Time response. Survival curves with data from nine separate experiments in which fungus was injected at different times after reconstitution of the lethally irradiated mice with 200 syngeneic HSCs and 500,000 culture-derived allogeneic mMPCs (bold gray) or 200 syngeneic HSCs without mMPCs (black). One group (Fungus day 4) received 660,000 mMPCs. The p values (log-rank analysis) are shown. mMPCs used in these experiments (Fig. 3A, B) were generated from C57BL6/Ka HSCs using a 7-day culture in the presence of IL-6.

Pooled cryopreserved mMPCs protect against fungal challenge in a dose-dependent manner. (A) Thirty-day survival of BALB/c mice challenged with Aspergillus fumigatus conidia 7 days after lethal irradiation and reconstitution with 200 syngeneic HSCs and 500,000 allogeneic culture-derived mMPCs from C57BL6/Ka mice, used either fresh or after cryopreservation. Data pooled from four experiments. mMPCs used in this experiment were generated by culturing for 7 days in the presence of IL-6. (B) Same as (A), but mice received escalating doses of cryopreserved allogeneic mMPCs, pooled from different strains (C57BL6/Ka, AKR, and FVB). Mice receiving HSCs and mMPCs (gray solid lines) had improved survival compared to mice receiving HSCs alone (black solid line). mMPCs used in these experiments were generated by 9-, 10-, and 10-day culture from C57BL6/Ka, AKR, and FVB HSCs, respectively. Survival at all mMPC doses was significantly improved compared to the group that received HSCs only. The p values shown in the graph indicate significance of differences between doses. Treatment groups receiving mMPCs consisted of 30 mice each. Gray dashed line represents mice receiving HSCs but no fungus, gray dotted line are vehicle controls exposed to radiation only. Statistical significance was assessed using Kaplan-Meyer survival analysis, with differences between groups analyzed by the log-rank test.
Almost all of the mMPC-derived cells seen at 4 weeks from day 8 mMPC or bone marrow MPs are long-lived B lymphocytes that likely originated from multipotent progenitors within the product but do not represent continuous de novo lymphopoiesis.
Allogeneic mMPC Product Protects Neutropenic Mice From Invasive Aspergillosis
The ability of allogeneic myeloid progenitors purified from bone marrow (a 1:2 mix of common myeloid progenitor and granulocyte macrophage progenitor) to protect neutropenic mice from fungal infection has been described previously (1,2). To determine whether ex vivo expanded mMPCs have the same potential, lethally irradiated mice (BALB/c, H-2d) were reconstituted on the same day with 200 syngeneic HSCs (c-Kit+Lineage-/low Sca-1+) and either 8 × 104 myeloid progenitors sorted as c-Kit+Lineage-/low Sca-1- cells from C57BL6/Ka, H-2b bone marrow (a mix of common myeloid progenitors, granulocyte-macrophage progenitors, and megakaryocyte-eryth-rocyte progenitors) or 5 × 105 mMPCs generated from HSCs after 7 days in culture. The mice were infected by tail vein injection with 150 conidia of Aspergillus fumigatus 7 days after the irradiation and cell transplantation. The experiment was repeated three times with 15 mice per group. As shown in Figure 3A, only 2/15 (13%) irradiation controls survived beyond 30 days, indicating that without treatment the radiation dose used was lethal to almost all mice. Injection of 200 HSCs at day 0 (HSC-rescue group) fully rescued the irradiated mice. However, only 17/44 (39%) of the HSC-only group survived subsequent injection of 150 Aspergillus fumigatus conidia, suggesting that HSC transplant alone does not generate sufficient myeloid cells in time to fully protect against fungal infection. Aspergillus was cultured from the spleen of infected animals that died, confirming invasive aspergillosis (data not shown). In contrast, 34/45 mice (76%) that received fresh bone marrow myeloid progenitors and 30/44 mice (66%) that received fresh, culture-derived mMPCs survived the fungal challenge. Statistical log-rank analysis showed significant protection by both bone marrow myeloid progenitor cells (p < 0.0001) and culture-derived mMPCs (p=0.0052) when compared to the HSCs-only group. There was no discernible difference in the 30-day survival between the groups that received bone marrow myeloid progenitors or culture-derived mMPCs (p = 0.26).
It has previously been shown that freshly isolated bone marrow myeloid progenitors did not protect from lethal fungal challenge until D+7 posttransplantation (2). Since mMPCs are a mixture of early progenitors and more differentiated cells, we tested how early posttransplantation mMPCs protected from Aspergillus infection (Fig. 3B). Recipient mice survived a fungal challenge administered during the initial day following irradiation, even in the absence of myeloid progenitors (data not shown), but became susceptible to infectious challenges administered on or after D+2. Unlike myeloid progenitors sorted from bone marrow (2) mMPCs expanded in culture protect against challenges administered as early as D+4 through to D+7. As reported previously (2), HSC-derived cells provide increasing levels of protection at day 10 and later.
Cryopreserved mMPCs Protect Allogeneic Neutropenic Mice From Aspergillosis in a Dose-Dependent Manner
To assess their functionality after cryopreservation, mMPCs cryopreserved after 7 days of culture were rapidly thawed in a 37°C water bath after at least 7 days of cryo-preservation and washed twice with medium. The recovered mMPCs were used to inject lethally irradiated allogeneic BALB/c hosts, which were challenged with Aspergillus fumigatus conidia as described for Figure 3. After fungal challenge in four independent experiments, there was no difference in survival between mice that received 500,000 fresh (59 mice total) or 500,000 cryopreserved (60 mice total) mMPCs (log-rank analysis, p = 0.47) (Fig. 4A).
mMPCs pooled from multiple allogeneic donor strains were also able to protect unmatched recipient mice from lethal fungal challenge in a dose-dependent manner (Fig. 4B). Following irradiation, BALB/c mice received 200 syngeneic HSCs or HSCs plus escalating doses of mMPCs pooled from C57BL6/Ka (H-2b), AKR (H-2k), and FVB (H-2q) mice cultured for 9, 10, and 10 days, respectively. The mice were infected with a lethal dose of Aspergillus fumigatus by intravenous injection on day 7 following radiation. Mice coinfused with mMPCs had significantly improved survival compared to mice receiving only HSCs. Survival of mice coinfused with mMPCs was mMPC dose dependent. At the 3 × 106 mMPC dose, 100% of mice were protected, significantly greater than the 20% survival observed in the cohort of mice receiving HSCs only (p < 0.0001). The deaths in the HSC+Aspergillus fumigatus group can clearly be attributed to infection, since all mice transplanted with HSCs but not inoculated with fungus survived.
Ex Vivo-Generated Myeloid Progenitors Engraft Across Allogeneic Barriers and Expand In Vivo
To demonstrate myeloid progenitor activity and assess the capacity of mMPCs to engraft across allogeneic barriers, 2.5 × 105 C57BL6/Ka mMPCs generated by 9-day cultures were transplanted into irradiated C57BL6/Ka or BALB/c mice. Day 8 and day 12 spleen colonies were formed in both recipient strains, demonstrating that mMPCs can engraft in syngeneic and allogeneic hosts (Fig. 5A). The spleen colonies generated in the allogeneic animals were fewer in number, approximately 0.5 colonies versus 1.5 colonies per 10,000 cells injected on day 12, and smaller than the resulting colonies in the syngeneic hosts. This result indicates that, while allogeneic mMPCs do face an engraftment barrier in fully ablated mice, they are able to engraft and proliferate to colonize the spleen.
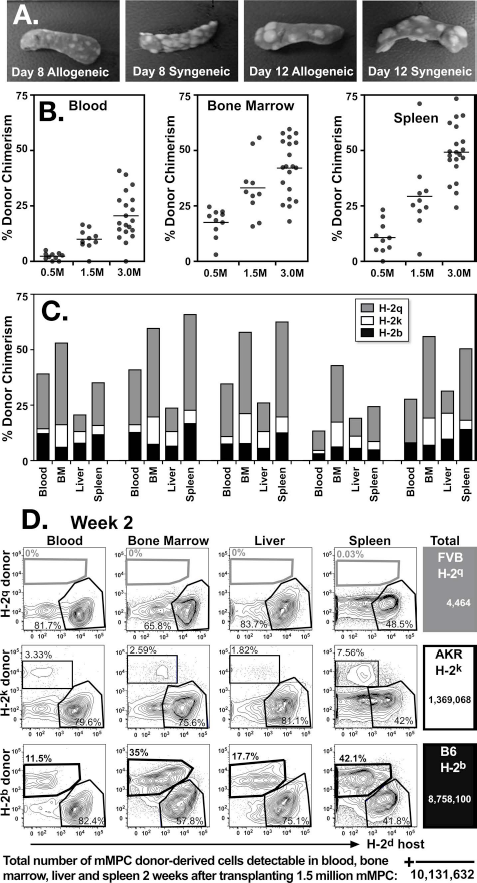
mMPCs provide protection from fungal challenge by engrafting across allogeneic barriers. (A) The 2.5 × 105 mMPCs derived from C57BL6/Ka were injected into syngeneic or allogeneic (BALB/c) recipients. mMPCs formed both day 8 and day 12 spleen colonies in syngeneic and allogeneic recipients. (B) Mice transplanted with 200 syngeneic HSCs and 5 × 105, 1.5 × 106, or 3 × 106 pooled allogeneic mMPCs (C57BL6/Ka, AKR, and FVB) were analyzed for total donor cell chimerism at 1 week posttransplantation. Total donor chimerism of the blood, bone marrow, liver, and spleen increased with escalating mMPC dose, (p < 0.0001, comparing 0.5 × 106 with 3 × 106 mMPCs in all organs). (C) The blood, bone marrow, liver, and spleen of recipient mice transplanted with 200 syngeneic HSCs and 3 × 106 pooled allogeneic mMPCs were analyzed by flow cytometry 1 week following transplantation for markers of mMPC donor HLA types (FVB, H-2q, gray; AKR, H-2k, white; C57BL6/Ka, H-2b, black). Shown are five mice from a representative experiment; contribution from all three mMPC donors was detectable in all mice. (D) Shown is an example of the in vivo expansion potential of mMPCs. BALB/c mice were lethally irradiated and transplanted with 200 syngeneic HSCs and 1.5 × 106 pooled allogeneic mMPCs (C57BL6/Ka, AKR, and FVB). The blood, bone marrow, liver, and spleen of recipient mice were analyzed 2 weeks following transplantation by flow cytometry for markers of host and mMPC donor HLA types. Flow cytometry plots show percentage of parental gates. Total donor cells in these tissues were calculated (right panel). mMPCs used in these experiments were generated by 9-, 10-, and 10-day culture from C57BL6/Ka, AKR, and FVB HSCs, respectively.
To determine the in vivo differentiation capabilities and localization of mMPC-derived progeny in mice post-transplantation, BALB/c mice were irradiated and transplanted with 200 BALB/c HSCs and 5 × 105, 1.5 × 106, or 3 × 106 previously cryopreserved mMPC derived from C57BL6/Ka, AKR, and FVB mice by 9-, 10-, and 10-day cultures, respectively. No infusion-related complications were observed. Blood, bone marrow, spleen, and liver were screened for mMPC donor cell MHC alleles. mMPC-derived chimerism in these tissues was dose dependent (Fig. 5B), and contribution from all three donor strains was detectable in all mice at week 1 (Fig. 5C).
In a selected mouse, the total number of donor cells present in the blood, bone marrow, liver, and spleen was determined 2 weeks after injection using flow cytometry and tissue counts (Fig. 5D). The engraftment kinetics of the three donor strains differed as the mMPC-derived progeny represented only two of the three original donors at this time point. FVB mMPCs (H-2q) provided the largest burst at week 1 and then decreased in frequency, while C57BL6/Ka (H-2b) dominated at week 2, illustrating the potential for donor variability in the kinetics of in vivo expansion (Fig. 5C, D). Greater than 10 million donor cells were enumerated in these tissues at week 2, which represents a 6.8-fold increase from the 1.5 × 106 mMPCs infused (Fig. 5D). This representative mouse had cells derived from C57BL6/Ka (H-2b) and AKR (H-2k) mMPCs at this time point. Of the total donor cells 8.7 × 106 were derived from C57BL6/Ka mMPCs, representing a 17.5-fold expansion from the 5 × 105 C57BL6/Ka mMPCs in pooled product at the time of infusion.
In Vivo Differentiation Potential of Ex Vivo-Expanded Myeloid Progenitor Cells
To determine the lineage potential of mMPCs in allogeneic irradiated recipients, donor progeny derived from the transplanted mMPCs were analyzed by flow cytometry 2 weeks posttransplantation (Fig. 6A). mMPC-de-rived CD11b+Gr-1-/low monocytes and macrophages and CD11b+Gr-1high granulocytes were detected in all tissues analyzed (Fig. 6B) (17). Erythroid progenitors, including CD71highTer119low proerythroblasts and CD71highTer119high and CD71low-medTer119high erythroblasts, were also evident (6). Donor dendritic cells, identified as MHC class 11+CD11c+, were detected in most mice. Donor-derived megakaryocytes were not present in quantities exceeding the detection limits in the tissues analyzed. T cells, which are not present in mMPC cultures, would not be expected to be generated from multipotent progenitor cells this early after HCT (7,36) and were not assessed in this experiment. mMPC-derived donor populations consisted primarily of myeloid, erythroid, and dendritic cell lineages 2 weeks after administration and less than 2% B220+ B lymphoid cells, except for one mouse with 4.6% B220+ donor cells in the bone marrow and one mouse with 3.3% B220+ donor cells in the blood (Fig. 6B).

In vivo differentiation of pooled allogeneic mMPCs. (A) BALB/c mice were transplanted with 200 syngeneic HSC and 1.5 × 106 pooled allogeneic mMPCs. Shown are representative FACS plots demonstrating the in vivo differentiation potential of mMPCs in the spleen 2 weeks postinjection. C57BL6/Ka donor cells (H-2b) were analyzed for the myeloid markers CD11b and Gr-1; CD11b+Gr-1-/low (monocytes and macrophages) and CD11b+Gr-1+ (granulocytes). CD11b-Gr-1- cells were further analyzed for markers of erythroid progenitors. Proerythroblasts and erythroblasts were distinguished with CD71 and Ter119. Donor dendritic cells (H-2b) were identified as MHC class II+CD11c+, both CD11b positive and negative dendritic cells were present. (B) BALB/c mice were lethally irradiated and transplanted with 200 syngeneic HSCs and 3 × 106 pooled allogeneic mMPCs. The blood, bone marrow, spleen, and liver of recipient mice were analyzed by flow cytometry 2 weeks following transplantation for markers of donor mMPC MHC class I types as well as for markers of myeloid (CD11b and Gr-1), erythroid (CD71 and Ter119), megakaryoid (CD41), dendritic (MHC class II, CD11c), and B-cell (B220) lineages. mMPCs gave rise to primarily erythroid, myeloid, and dendritic cell types. Donor chimerism determined by flow cytometry as percentage of live cells. mMPCs used in (A) and (B) were generated by 9-, 10-, and 10-day culture from C57BL6/Ka, AKR, and FVB HSCs, respectively. (C) BALB/c mice were transplanted with 200 syngeneic HSCs and 1.5 × 106 mMPCs derived from C57BL6/Ka EGFP+ mice. Shown is a representative mouse bled at 7, 14, and 21 days posttransplantation. Donor platelets were identified by expression of CD41, CD61, and EGFP. Numbers indicate the percentage of parental population.
To determine if mMPCs also gave rise to platelets in vivo, mice expressing EGFP under the β-actin promoter in all cells including platelets (C57BL6/Ka EGFP+) were utilized to generate mMPCs for transplantation (38). BALB/c mice were irradiated and transplanted with 200 syngeneic HSCs and 1.5 × 106 allogeneic EGFP+ mMPCs. EGFP+ platelets were present in the blood of transplanted mice, peaking 2 weeks posttransplantation (Fig. 6C).
mMPC Engraftment Is Transient
mMPCs derived from C57BL6/Ka, AKR, and FVB mice by culture for 9-, 10-, and 10-day cultures, respectively, engraft and expand rapidly in BALB/c mice as shown in Figure 5; however, engraftment is transient (Fig. 7A). In syngeneic HSC transplanted mice, the bone marrow and spleen cellularity increase steadily for several weeks after HCT and eventually return to normal values (13,36). Conversely, the contribution of mMPC-derived donor cells peaks at 1 to 2 weeks posttransplant and is mostly undetectable 4 weeks after HCT.

mMPC engraftment is transient. (A) BALB/c mice were lethally irradiated and transplanted with 200 syngeneic HSCs and 3 × 106 pooled allogeneic mMPCs. Bone marrow and spleen were analyzed 1, 2, and 4 weeks posttransplantation for total cell count and mMPC donor-derived cell counts. Donor chimerism was determined by flow cytometry as percentage of live cells and multiplied by total tissue cellularity to determine the donor cell count. Each data point represents an individual mouse. (B) The blood of BALB/c mice that had received pooled allogeneic mMPCs and had survived a fungal challenge was analyzed for mMPC-derived (donor) chimerism. Shown is mMPC-derived chimerism 12 weeks posttransplantation at the 3 × 106 cell dose. Three of 30 mice have low donor chimerism, consisting primarily of lymphoid-only chimerism, characteristic of the presence of few multipotent progenitors in the cultured cells. Data were combined from two experiments. mMPCs used in these experiments were generated by 9-, 10-, and 10-day culture from C57BL6/Ka, AKR, and FVB HSCs, respectively.
To assess the persistence of mMPC-derived cells, mice that had been reconstituted with allogeneic mMPCs (and syngeneic HSCs) and had survived a fungal challenge (Fig. 4B) were screened for the presence of mMPC-derived cells by flow cytometry using antibodies against myeloid, T-, and B-cell lineages. Blood analysis of the surviving mice 12 weeks posttransplantation revealed that circulating mMPC-derived donor cells were no longer present in most mice analyzed, demonstrating that mMPC engraftment was generally transient (Fig. 7B). Similar results were obtained 20 weeks posttransplantation (data not shown). The persistent low-level donor lymphoid contribution (<10%) in a small percentage of mice is likely due to the presence of a few transiently engrafting multipotent cells remaining in the cultures. These lymphoid cells are derived from progenitors in the transplanted mMPCs as no detectable lymphoid cell or lymphoid progenitor cells are present in the mMPC product itself. These progenitors have to undergo selection in the host environment (35); hence, the lymphoid cells generated are not expected to induce graft versus host disease (GvHD). Indeed, no signs of GvHD have been observed in this model even following infusion of high mMPC doses.
mMPC Treatment Does Not Lead to Detectable Alloimmunization
Alloimmunization from mMPC treatment is a concern even following transient engraftment as it may increase the likelihood of failure of subsequent allogeneic bone marrow transplantation (BMT). Recent research has indicated that alloimmunization results in the presence of circulating antibody and is the major barrier to engraftment in BMT (34). To determine if mMPC-treated mice become alloimmunized, we transplanted lethally irradiated BALB/c mice with 200 syngeneic HSCs or syngeneic HSCs in combination with 3 × 106 pooled mMPCs derived from C576BL6/Ka, AKR, and FVB. Fully immunocompetent mice were primed by intraperitoneal injection of 20 × 106 splenocytes from C57BL6/Ka or AKR mice to serve as positive controls for alloimmunization. The mMPC-transplanted and the splenocyte-primed mice were lethally irradiated 6 weeks later and infused with 20 × 106 C57BL6/Ka EGFP+ T-cell-depleted bone marrow. The bone marrow and spleens of mice were analyzed by flow cytometry 3 days following infusion of the C57BL6/Ka-EGFP+ cells for the presence of transplanted cells. C57BL6/Ka-EGFP+ cells were detected in all mice originally transplanted with HSCs (4/4) or HSCs plus mMPCs (8/8) (Fig. 8A), indicating that allosensitization toward C57BL6/Ka cells had not occurred. All mice primed with C57BL6/Ka splenocytes (10/10) showed no EGFP, indicating complete rejection of the C57BL6/Ka-EGFP+ graft. Of the mice primed with AKR splenocytes, fewer than half (4/10) had very low numbers of C57BL6/Ka-EGFP+ cells, confirming that a cross-reactive antibody response can occur during alloimmunization (34).
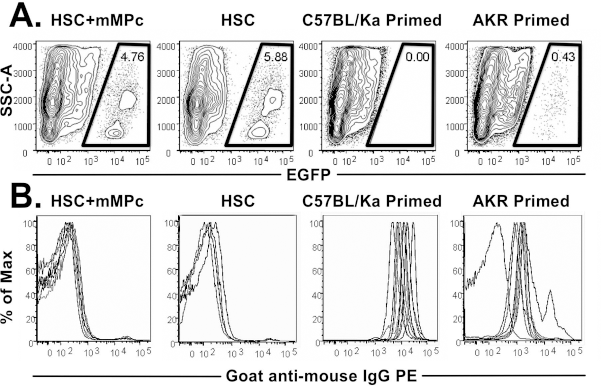
mMPC engraftment does not lead to detectable alloimmunization. (A) BALB/c mice were initially transplanted with 200 syngeneic HSCs or syngeneic HSCs and 3 × 106 pooled allogeneic mMPCs. Positive control mice were primed with 20 × 106 AKR or C57BL6/Ka splenocytes. Six weeks after transplantation or priming mice were irradiated and infused with 20 × 106 C57BL6/Ka EGFP+ T-cell-depleted bone marrow cells. Three days later, mice were analyzed for the presence of EGFP+ cells. All mice transplanted with HSCs or HSCs and mMPCs had detectable EGFP+ cells. Mice primed with 20 × 106 AKR or C57BL6/Ka splenocytes were alloimmunized and rejected the EGFP+ bone marrow. Shown are representative flow cytometry plots. (B) Serum was collected from the above mice to screen for allo-antibodies capable of binding C57BL6/Ka thymocytes. All mice originally transplanted with syngeneic HSCs (mean fluorescence intensity 336.5 ± 37.51) or syngeneic HSCs and mMPCs (mean fluorescence intensity 355.25 ± 77.53) did not have alloreactive serum antibodies (p = 0.5848). Mice primed with C57BL6/Ka (mean 15,779.6) or AKR (mean 2,267.6) splenocytes had serum alloreactive antibodies. Shown are cells stained with a 1:10 dilution of serum. Histograms representing all mice in each group are shown (n = 10, except “HSC” group, where n = 8; different line for each mouse). Statistical significance was evaluated using unpaired t-test. mMPCs used in these experiments were generated by 9-, 10-, and 10-day culture from C57BL6/Ka, AKR, and FVB HSCs, respectively.
Serum was collected from all mice in the alloimmunization study to screen for the presence of antidonor antibody. Serum was diluted and incubated with C57BL6/Ka thymo-cytes for 1 h on ice. After washing, the cells were incubated with a PE-conjugated goat anti-mouse IgG and analyzed by flow cytometry to detect bound mouse antibody. No difference in fluorescence intensity was observed between the mice transplanted with syngeneic HSCs or HSCs plus mMPCs (Fig. 8B). The mice primed with C57BL6/Ka or AKR splenocytes, on the other hand, had antidonor anti-bodies. The intensity of antibodies recognizing antigens on C57BL6/Ka cells in the mice primed with AKR cells was lower than in mice primed with C57BL6/Ka cells, consistent with the clearance data.
Discussion
Despite the use of broad-spectrum antimicrobials and colony-stimulating factors, severe neutropenia continues to be an important clinical problem, putting patients at risk of life-threatening infections and reducing or delaying chemotherapy treatments (14,19). Up to 20% of HCT patients contract invasive fungal infections, the majority being Aspergillus (15,31). Mortality in these patients can be as high as 40-90% depending on underlying patient risk factors, highlighting the need for new innovative therapies (12,27). Here we demonstrate that ex vivo culture-derived allogeneic myeloid progenitors can expand and differentiate in vivo into myeloery throid cells, including functional granulocytes. Culture conditions were empirically selected to maximize the myeloid progenitor content, without generating large quantities of mature cells. Therefore, we focused on cytokine combinations that emphasize proliferation over differentiation. In addition clinical translation (defined conditions and reagents suitable for clinical manufacturing) was a consideration. Our data establishes that cryopreserved mMPCs are effective in preventing death from invasive fungal infection upon thaw.
Several different Aspergillus infection models exist, including different routes of administration, and different modes of inducing neutropenia. While intranasal inoculation may be considered more physiological, an intravenous injection model was chosen for these initial studies because of the reduced variability in tissue distribution associated with intravenous compared to intranasal injection. Similarly, whole body irradiation results in a more reproducible period of neutropenia that is also more severe and prolonged when compared to chemotherapy induced. Moreover, we have demonstrated the protective benefit of myeloid progenitors following both chemotherapy and intranasal instillation of fungus (2,3).
Studies of granulocyte transfusion therapy have established that large numbers of allogeneic myeloid cells can be safely infused, are functional, migrate to sites of infection, and are not immediately cleared by the host's immune system (11,16,29). However, determining the utility of granulocyte transfusions for ongoing infections during neutropenia has been confounded by lack of technologic advances and the paucity of randomized controlled trials (20,24,28). The results of a recently completed, randomized, controlled, NIH-sponsored trial of granulocytes mobilized by G-CSF are much anticipated (NCT00627393; clinicaltrials.gov). Nevertheless, these transfusions present tremendous practical challenges for the patients, donors, and transfusion services due to the fact that these cells cannot be cryopreserved and need to be infused frequently.
Our data support the numerous immunologic and practical advantages of a standardized allogeneic human myeloid progenitor product over granulocyte transfusions. Given the expansion potential of mMPCs in vivo it is expected that substantially fewer human MPCs relative to mature granulocytes would need to be infused for efficacy, especially since the expansion numbers seen underestimate the total number of cells generated as cells continuously mature, enter the bloodstream and pass into the tissues, or conclude their lifespan. Lower transfusion numbers reduce the probability of infusion-related complications. The greatest hindrance in the use of granulocyte transfusions is the inability to store mature granu-locytes in a functional state (28). Also, donors, apheresis equipment, and personnel need to be available to collect granulocytes for prompt infusion. Moreover, mature granulocytes have a short life span in the blood requiring almost daily administration of large numbers of cells. In contrast, mMPCs provide rapid production of functional granulocytes. Engraftment is transient but sustained the diverse types of progenitors contained in mMPC over the period of several weeks posttransplant when neutropenia is most pronounced. These diverse progenitor cell types include myeloid progenitors and committed precursors as well as phenotypic Lineage-/lowc-Kit+Sca-1+ hematopoietic stem and multipotent progenitor cells. Our data suggests that these Lineage-/lowc-Kit+Sca-1+ cells include multipotent progenitors with high potential to generate myeloid progeny for a period of several weeks, but limited self-renewal capacity. Hence their engraftment is predominantly transient in an allogeneic transplant scenario and can be influenced by culture duration. Because lymphoid progenitors and mature lymphoid cells are not present in mMPCs and the few observed T cells derived from the stem and multipotent progenitor cells will have undergone thymic education in the host after transplantation (35), no signs of GvHD are expected, nor have they been observed.
A further benefit of MPCs over granulocyte transfusion is that they can be cryopreserved, probably for many years without loss of function (4,5). Ongoing stability studies with human MPCs have shown no loss of activity for up to 3 years of cryopreservation as determined by an in vitro potency assay (data not shown). Therefore, a myeloid progenitor product could be available as needed at any medical facility, avoiding the potential delays in administration associated with granulocyte transfusions. In addition to neutrophils, mMPCs also produce platelets in vivo. Platelets have been shown to attach to cell walls of invasive hyphae of Aspergillus fumigatus, augmenting neutrophil-mediated effects (8).
Aside from infusion-related toxicities, rejection and alloimmunization are a concern following transfusion of blood products. Infusion of allogeneic granulocytes into neutropenic patients results in increases in the absolute neutrophil count in the blood that is still measurable 24 h later, indicating these cells are not rapidly cleared (16). While a minor engraftment barrier was observed for allogeneic mMPCs in immunocompromised mice as shown by CFU-S, the barrier did not compromise overall efficacy. Administration of mMPCs not only increased circulating WBC counts 1 week after administration but also improved hematopoietic tissue cellularity at this time point compared to mice that did not receive mMPCs (data not shown). Therefore, an allo-barrier is not expected to significantly affect the efficacy of a human myeloid progenitor product developed for use in immunocompromised patients. Rapid alloimmunization from multiple granulocyte transfusions has not been routinely observed (10,29). In the presented studies, conditioned mice transplanted with mMPCs also did not generate antidonor anti-bodies. It is therefore expected that a human allogeneic myeloid progenitor product could be safely administered with a low risk of alloimmunization.
A few small studies have attempted to reduce infections following HCT by infusion of ex vivo-expanded autologous stem and progenitor cells. In the majority of these studies improvement in time to neutrophil engraftment (21,25,26) and a reduction in neutropenic fever (30) were observed compared to historic controls. A universal, allogeneic myeloid progenitor product as presented here would be superior to expanded autologous cells and, more importantly, have far broader applicability for patients with neutropenia. Central, large-scale production of pooled allogeneic MPCs would ensure a readily available product at reduced cost, without the logistical challenges and risks of autologous cell transplants or granulocyte transfusions. Pooling would also minimize the variability, evident from the engraftment data presented here, encountered between different donors, resulting in a more reproducible MPC product. CTL-008, currently in Phase 2 clinical trials in acute leukemia patients with chemotherapy-induced neutropenia, may be an effective bridging therapy able to provide a rapid and sustained supply of functional granulocytes to reduce infectious sequelae in patients with neutropenia.
Footnotes
Acknowledgments
We thank Mastura Wahedi, Esther Danenberg, and Myrrh Sagy for excellent technical assistance in performing these experiments. We thank Andrew Bitmansour for help in setting up the infection assays, and we thank Holger Karsunky, Elisa Brunette, Margaret Dillon, Debbie Strachan, Madhavi Anumula, Gregory Boucher, and Kimberly Gandy for critically reviewing the manuscript. This work was supported by the National Institute of Allergy and Infectious Diseases (1 R43 A1064156-01 and 1R43 A1064156-02). All authors were employed by Cellerant Therapeutics, Inc., at the time this work was performed, except J.M.Y.B., who is a consultant to the company.