
Abstract
Stem cell transplantation is a viable strategy for regenerative medicine. However, it is inevitable to have cells undergo storage for several hours or days due to processing and transportation. Therefore, it is crucial to have rigidly controlled conditions ensuring the therapeutic benefit of isolated stem cells. In the present study, we investigated the impact of short-term storage on human CD133+ cells. CD133+ cells were isolated from human bone marrow and kept at standardized nonfreezing storage conditions for up to 72 h. Cell viability (apoptosis/necrosis) and expression of CD133 and CXCR4 were analyzed by flow cytometry. Metabolic activity was determined using an MTT assay; colony-forming ability, as well as endothelial-like differentiation, was further evaluated. A qRT-PCR array was employed to investigate the expression of sternness genes. CD133 and CXCR4 expressions were preserved at all time points. After 30 h, cell number and metabolic activity decreased, although no significant changes were detected in cell viability and proliferation as well as endothelial-like differentiation. Cell viability and proliferation decreased significantly only after 72 h of storage. Our results indicate that storage of isolated human CD133+ bone marrow stem cells in liquid allows for high viability and functionality. However, storage time should be limited in order to avoid cell loss.
Introduction
As a means of restoring the function of ischemic heart tissue, stem cell transplantation has received much scientific attention over the last decade. Following the first report about beneficial effects of bone marrow (BM) cells in an animal model of myocardial infarction (29), the potential of bone marrow mononuclear cells (BMMNCs) as well as isolated stem cells was investigated intensively (15,24,44). Immunomagnetic cell separation devices allow fast and sterile preparation of autologous stem cells. Currently, more than 80 clinical trials concerning the application of stem cells to enhance heart function are registered (27). The safety and efficacy of BM CD133+ stem cell transplantation into the heart has been shown in our group (37,38). An ongoing phase III clinical trial has been designed to further investigate the therapeutic impact of stem cell transplantation during coronary artery bypass graft surgery.
For multicenter trials, the comparability of cell product quality for all patients enrolled is of supreme importance. It may be ensured by tightly controlled transport processes, which necessitate storage of aspirated BM as well as isolated stem cells. In a clinical setting, autologous stem cells may be transplanted 24-30 h after BM aspiration. Conditions of storage for optimal cell quality preservation are therefore essential, and their importance will increase once stem cell therapy evolves to be a routine pharmaceutical treatment.
Short-term storage of BM samples at 4°C is used in several clinical studies (39,40,47). However, a reduction in progenitor cell number and proliferation occurs according to Kao et al. (17). This group reported a CD34+ cell recovery of 75% after 72 h of storage and a linear reduction of the total number of colonies formed in a colony-forming unit (CFU) assay, reaching approximately 30% after 72 h.
Clinical trials using BMMNCs such as ASTAMI, REPAIR-AMI, and others employ cellular products stored for 1 day and confirm high viability of these cells (23,34). However, whether the use of a homogenous, freshly isolated stem cell population may carry significant advantages for therapy and/or cell product stability remains an open question.
BMMNCs showed reduced invasion capacity after overnight storage in saline with 20% serum (35). The expression of CXCR4, a surface receptor that binds the chemoattractant stromal cell-derived factor 1 (SDF-1) and thus contributes to chemotaxis, was significantly downregulated after storage. As CXCR4 expression and Chemotaxis in response to SDF-1 are not restricted to stem cells, the importance of Seegers et al.'s (35) findings in relation to hematopoietic stem cells (HSCs) needs to be further clarified.
The impact of short-term storage on viability and proliferative capacity of isolated CD133+ HSCs has not been systematically addressed so far. In the present study, we analyzed viability, colony-forming capability (CFU-EC), metabolic activity, and stemness marker expression of freshly isolated compared to stored CD133+ stem cells derived from BM.
Materials and Methods
Bone Marrow Aspiration
BM was aspirated from informed donors who gave written consent to the use of their BM for research according to the Declaration of Helsinki. The ethical committee of the University of Rostock has approved the presented study (registered as No. A 2010 23) as of April 29, 2010. BM samples were obtained by sternal aspiration from patients undergoing coronary artery bypass graft surgery at Rostock University Hospital, Rostock, Germany. Samples of 36 donors were included; mean volume was 49.8 ± 2.7 ml, and anticoagulation was achieved by heparinization with 250 IU/ml sodium heparin (B. Braun Melsungen GmbH, Melsungen, Germany). Mean donor age was 66.8 ±2.1 years, and 80.6±% of the donors were male.
Cell Isolation and Storage
MNCs from all samples were isolated by density gradient centrifugation on 1077 Lymphocyte Separation Medium (LSM; PAA, Pasching, Austria). CD133+ cells were enriched by positive magnetic selection using the MACS cell separation system (Miltenyi Biotec, Bergisch Gladbach, Germany). Purity and viability of all cell isolations were verified by flow cytometry. Autologous plasma was prepared from a small portion of the BM sample and used for cell storage [10% autologous plasma in 0.9% saline (B. Braun Melsungen)]. Trypan blue (concentration 0.4%; Sigma-Aldrich, St. Louis, MO, USA) was used for live/dead cell counting in a Neubauer hemacytometer (Carl Roth, Karlsruhe, Germany). Based on clinical cell processing, cells were counted before static storage at 4°C, and equal volumes of cell suspension were used for each time point.
Fluorescence-Activated Cell Sorting Analysis (FACS) for Surface Markers
The following antibodies were used for flow cytometric characterization: anti-CD133-PE (293C2, 50 μg/ml, 1:10 dilution), isotype control mouse IgG 2b-PE (1:10 dilution) and anti-CD34-FITC (AC136, 1:10 dilution) (Miltenyi Biotec), anti-CXCR4-APC (12G5, 1:5 dilution; Becton Dickinson, Franklin Lakes, NJ, USA), and isotype control mouse IgG 2a κ-APC (1:5 dilution; BioLegend, San Diego, CA, USA).
Cells were suspended in phosphate-buffered saline (PBS; PAN-Biotech GmbH, Aidenbach, Germany) containing 2 mM EDTA (Sigma-Aldrich) and 0.5% bovine serum albumin (Sigma-Aldrich), and FcR blocking reagent (Miltenyi Biotec) was employed to reduce unspecific binding. Antibodies were added to the cell suspension and incubated for 10 min in the dark at 4°C. Cells were washed with cold PBS and analyzed by LSR-II flow cytometer (Becton Dickinson). Data analysis was performed with FACSDiva software (Becton Dickinson). Boolean gating was performed in three steps:
Selection of cells according to FSC/SSC as shown in Figure 1A
Selection of viable (7-AAD negative) cells as shown in Figure 1B
Analysis of viable cell CD133/CXCR4 expression

Characterization of freshly isolated CD133+ cells. (A) CD133+ cells isolated from BM by magnetic selection and analyzed by flow cytometry formed a population of characteristic morphology in a forward scatter area (FSC-A) versus sideward scatter area (SSC-A) dot plot. (B) Sample plot of cells stained with 7-AAD. (C, D) Overlay plot of freshly isolated cells stained with antibodies (white) and appropriate isotype controls (gray); 90.5% of cells were positive for CD133 (C), 11.2% expressed CXCR4 (D). (E) Confocal microscopy of CD133+ HSCs showed CD34 to be distributed evenly on the cell surface (green), whereas CD133 was restricted to distinct foci (red). Representative x-y sections from a z stack are shown (upper panel: fluorescence channel overlay, lower panel: corresponding bright field image; z values are presented in the lower left corner of each image). Scale bar: 10 μm.
Apoptosis/Necrosis Assay
A FITC Annexin V apoptosis detection kit and 7-AAD were purchased from Becton Dickinson and used according to the manufacturer s instructions. Cells were washed in PBS prior to staining and analyzed by LSR-II flow cytometer with FACSDiva software.
MTT Assay
Metabolic activity of living cells was determined by MTT assay as follows.
Freshly isolated CD133+ cells were counted, and equal amounts of cell suspension were subjected to the assay at the indicated time points. Cells were prewarmed in StemSpan H3000 (Stemcell Technologies Inc., Vancouver, BC, Canada) supplemented with interleukin-3 (IL-3) and stem cell factor (SCF, 2 ng/ml each; PAN-Biotech) for 30 min. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (Carl Roth) was added to a final concentration of 500 μg/ml and cells were incubated for 4 h. Absorption was determined at 550 nm and 655 nm (reference wavelength).
Colony-Forming Unit-Endothelial Cell (CFU-EC) Assay
CD133+ cells form colonies in methylcellulose gel containing suitable cytokines and have been shown to differentiate to endothelial-like cells in vitro (28). CFU-EC was performed as previously described (28). In brief, cells were embedded in semisolid medium (composition: see Table 1).
Composition of Semisolid Medium for CFU-EC
Colonies were counted after 26 days, and gels were dissolved for further use of differentiated cells. For immunofluorescence, differentiated cells were plated on fibronectin-coated coverslips [Human Plasma Fibronectin from Merck Millipore, Billerica, MA, USA; 10 μg/ml in PBS, coated for 1 h at room temperature (RT)], and cultivated in EGM-2 (Lonza Group Ltd., Basel, Switzerland) for 3 days.
Immunofluorescence and Uptake of Acetylated LDL
Freshly isolated CD133+ cells were incubated with anti-CD133-PE (293C2, 50 μg/ml, 1:10 dilution) and anti-CD34-FITC (1:10 dilution, Miltenyi Biotec), washed, and directly visualized at RT. Endothelial-like cells that differentiated from CD133+ cells were incubated with 10 μg/ml Alexa Fluor 488-labeled acetylated LDL (Life Technologies, Grand Island, NY, USA) for 1 h at 37°C and 5% CO2. Cells were washed with PBS and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 6 min at RT. To visualize intracellular von Willebrand factor (vWF), cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich) in PBS, and unspecific binding of antibodies was blocked by incubation with 3% bovine serum albumin (Sigma-Aldrich) for 1 h at RT. Rabbit anti-vWF (H-300, 200 μg/ml, employed 1:100 in PBS overnight at 4°C) was procured from Santa Cruz Biotechnology (Dallas, TX, USA) and donkey anti-rabbit-Alexa Fluor 488 (2 mg/ml, used 1:350 for 3 h at 37°C) from Life Technologies. Staining with 300 nM 4′-6-diamidino-2-phenylindole (DAPI, Life Technologies) in PBS for 5 min at RT was used as a counterstain for nuclei. Fixed and stained cells were washed extensively and mounted in FluorSave (Merck Millipore).
Microscopy was performed on a LSM780 ELYRA/PS-1 (Carl Zeiss AG, Jena, Germany) in LSM mode using ECPlanNeoFluor 40x/1.3 Oil DIC objective with immersion oil (Immersol™ 518F; Carl Zeiss). Excitation of Alexa Fluor 488 as well as FITC was achieved by 488 nm laser and recorded at 498-561 nm emission [main dichroic beam splitter (MBS 488/561)], a 561-nm laser was employed to excite PE detected at 567-647 nm (MBS 488/561), and DAPI was excited by a 405-nm laser and recorded with a 415- to 470-nm emission filter (MBS 405). Transmitted light images were collected simultaneously with the excitation of DAPI by using a photomultiplier for transmitted light. The presented photomicrographs were generated with ZEN software (Carl Zeiss AG).
Stemness Gene qRT-PCR Microarray
Freshly isolated or stored CD133+ cells were washed with PBS, 2 mM EDTA, and 0.5% bovine serum albumin. The cell pellet was snap frozen and stored in the vapor phase of a liquid nitrogen tank. Cell pellets were thawed on ice, and total RNA was isolated by RNEasy Micro Kit (Qiagen GmbH, Düsseldorf, Germany). Reverse transcription was performed using SuperScript II reverse transcriptase kit (Life Technologies). An aliquot of cDNA generated from 100 ng RNA was employed as template per 96-well plate. A StellArray Embryonic Stemness Gene qRT-PCR kit (Lonza) was used according to the manufacturer s instructions.
Statistical Analysis
Statistical analysis was performed with SigmaPlot 11.0 (Systat Software, Inc., San Jose, CA, USA). Data are presented as mean ± SD unless indicated otherwise. Continuous variables were tested for normal distribution with the Shapiro–Wilk test. For normally distributed variables, groups were compared by repeated-measures one-way ANOVA. The Friedman repeated measures analysis of variance on ranks was applied for non-normally distributed variables. Post hoc tests and pairwise multiple comparisons were performed with the method of Holm–Sidak for normally distributed variables. A probability value of p < 0.05 was considered to denote statistical significance.
Results
Hematopoietic Stem Cells Selected for CD133 Were Viable After 30 h of Storage, but Were Subject to a Significant Decrease in Viability After 72 h of Storage
Quality of freshly isolated CD133+ stem cells from human BM samples was ascertained by flow cytometry. Samples included in this study showed a CD133 expression of more than 70% and a minimum viability, as assessed by 7-AAD exclusion, of 85%. Flow cytometric parameters of a representative sample of freshly isolated CD133+ cells are depicted in Figure 1A-D. Confocal immunofluorescence microscopy revealed typical characteristics of CD133+ HSCs: cells were evenly shaped and had a diameter ≤10 μm; CD34 was evenly distributed over the cell surface, whereas CD133 was present only in distinct foci of the plasma membrane (Fig. 1E). Morphological characteristics, as well as marker distribution, were in accordance with data published previously by Giebel et al., further confirming cell type and quality (13). The mean cell density of isolated cells throughout liquid storage was 4.60 × 106/ml.
Freshly isolated cells were 17.2 ± 7.5% annexin positive, but 7-AAD negative and were therefore described as apoptotic (Fig. 2A, B). The percentage of apoptotic cells increased slightly to 19.0 ± 10.9% after 30 h of storage and 24.6 ± 12.7% after 72 h. However, the differences were not significant. Similarly, no significant rise of the necrotic cell population was detected. Necrotic cells, including late stage apoptotic cells, were defined as 7-AAD positive annexin positive/negative and occurred at a frequency of 12.1% in freshly isolated cells.

Cell number and viability decreased significantly only after 72 h. (A) Flow cytometry plots of 7-AAD/annexin-FITC staining for evaluation of early apoptosis (annexin positive, 7-AAD negative) and late apoptosis/necrosis (7-AAD/annexin double positive or 7-AAD positive). (B) Percentage of apoptosis and necrosis. Black bar, apoptotic cells and light gray bar, necrotic cells. n = 7. (C) Changes in cell number over time (Neubauer hemacytometer count). n = 7. (D) Cell viability as assessed by trypan blue exclusion. n = 7, *p < 0.05.
Total cell number decreased significantly over storage time (4.00 ± 1.14 × 106/ml at 0 h, 2.65 ± 0.96 × 106/ml after 30 h, and 2.46 ± 1.12 × 106 cells/ml after 72 h) (Fig. 2C). Viability, evaluated by trypan blue exclusion (Fig. 2D), decreased from 88.2 ±9.0 (0 h) to 79.4 ± 10.1 (30 h) and 63.9 ± 17.6 (72 h); again, the viability loss after 72 h was significant compared to 0 h.
CD133+ HSCs Proliferate and Differentiate After Storage
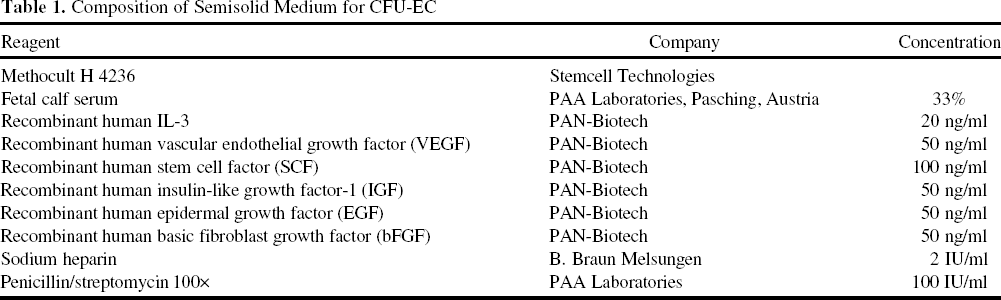
The effectiveness of stem cells for clinical therapy relies on cellular functions including the ability to proliferate, either for self-renewal or for differentiation into mature, specialized, and functionally active cells. Therefore, we investigated the potential of CD133+ HSCs to proliferate and generate offspring with endothelial-like properties using the CFU-EC assay (28). On average, 285.8±212.1 colonies were formed by 1,000 freshly isolated CD133+ cells (Fig. 3A). Cells seeded after storage for 30 h generated 297.6 ± 317.6 colonies. Thus, proliferation was not significantly influenced after 30 h of storage. In contrast, a striking reduction of colony-forming ability was observed after 72 h of storage (23.2 ± 27.9 colonies, p < 0.05 compared to fresh or to 30 h). Colony morphology remained unchanged (Fig. 3B).
Proliferation of cells was stable after 30 h of storage but sharply decreased after 72 h. (A). Colonies formed in CFU-EC are depicted as number of colonies per 1,000 cells. n = 7, *p < 0.05. Colony morphology remained stable over time (B). Representative microphotographs; scale bar: 100 μm.
Cells isolated from CFU-EC colonies were further subjected to cultivation on fibronectin-coated coverslips for 3 days. Progeny of cells stored for 72 h could not be cultivated and analyzed due to low colony numbers. Adherent cells were confirmed to possess endothelial features as they incorporated acetylated LDL labeled with Alexa Fluor 488 (Fig. 4A) and expressed vWF, which was detected by immunofluorescence (Fig. 4B).

Immunophenotyping of endothelial markers in endothelial-like cells derived from CD133+ cells. (A) Uptake of acetylated LDL labeled with Alexa Fluor 488 (acLDL AF488) into cells differentiated from freshly isolated CD133+ cells (upper panel) or from CD133+ cells stored for 30 h (lower panel). Nuclei were stained with DAPI. Morphology of cells as detected by transmitted light photomultiplier is shown on the right (phase contrast). Scale bar: 10 μm. (B) Expression of vWF in cells differentiated from freshly isolated CD133+ cells (upper panel) or from CD133+ cells stored for 30 h (lower panel). Control staining (secondary antibody only) is shown on the right. Nuclei were stained with DAPI. Scale bar: 10 μm.
A Significant Decrease in Metabolic Activity Preceded the Loss of Proliferation Capacity
The metabolic activity of freshly isolated and stored CD133+ cells was investigated via MTT assay. After 30 h and 72 h of storage, cellular metabolic rate dropped to 38.3 ± 10.9% and 39.2 ± 39.48% compared to the metabolism in fresh cells (Fig. 5A). The decrease was significant for both time points.

Metabolic activity of cells decreased after 30 h of storage, whereas CXCR4+CD133+ cell population did not change significantly over time. (A) Metabolic activity. Results were normalized to fresh cells. n = 6, *p < 0.05. (B-E) CD133/CXCR4 expression on viable cells over time. Plots display median (line) with 25% to 75% interquartile range (box). Despite a trend toward CD133 loss, none of the populations changed significantly. n = 8.
CXCR4 Expression Was Stable for Up to 72 h of Storage, CD133 Expression Decreased Insignificantly
As a marker of stemness that is lost early on during in vitro culture, CD133 was analyzed before and after storage. CXCR4, receptor of the HSC-related chemokine SDF-1, which has been detected in ischemic myocardium (1), was determined likewise (Fig. 5B-E). Subpopulation analysis revealed a tendency of CD133 protein loss and a possible shift of CXCR4+ cells from the CD133+ to the CD133- cell population. After storage for 72 h, the double-negative cell population displayed a trend to increase, while viable cells coexpressing CD133 and CXCR4 tended to decrease. However, these changes did not reach the level of significance. In median, 52.3% of freshly isolated viable cells coexpressed CD133 and CXCR4. Total CD133+ expression was 84.0±5.9% on living cells immediately after isolation. A decrease during storage proved insignificant (69.2 ± 26.7% after 30 h of storage, 64.5 ± 20.5% after 72 h).
CXCR4 expression remained stable on total viable cells (0 h: median 66.35%, 30 h: median 70.60%, 72 h: median 72.65%), as well as on viable CD133+ cells (0 h: median 64.74%, 30 h: median 70.92%, 72 h: median 70.91%). Flow cytometric sample plots of single staining and isotype controls illustrate the employed gating strategy (Fig. 6A). Sample plots of multicolor analyses at the chosen time points are shown in Figure 6B.

Multicolor flow cytometric analysis (7-AAD/CD133/CXCR4) based on Boolean gating. (A) Single staining of CD133 and CXCR4 antibodies as well as isotype controls were employed for gating. (B) Representative plots of multicolor staining at different time points.
CD133+ HSCs Expressed Stemness Genes After Storage
Transcriptome analysis of cells from three donors demonstrated the expression of six stemness-related markers in freshly isolated CD133+cells, five of which were retained in all samples stored for 30 h (Table 2). FGF2 was detectable in two of three stored samples. None of the markers were affected significantly.
Stemness-Related Genes Identified by qRT-PCR
Discussion
HSCs are increasingly used in regenerative medicine. In order to facilitate multicenter studies encompassing large numbers of patients carefully selected by extensive inclusion criteria, cell preparations need to be stored and transported for up to several days. In the present work, we investigated the impact of short-term nonfreezing storage on purified BM-derived CD133+ HSCs. We found that (i) cell viability and colony-forming capability remained high up to 30 h of storage, but decreased after 72 h of storage. Cell number and metabolic activity (ii) were significantly higher in freshly isolated cell product compared to stored cells. The rate of (iii) apoptotic and necrotic cells remained constant throughout all storage time points. Finally, we found the (iv) expression of stem cell markers on isolated CD133+ cells and endothelial markers after colony-forming assay was not changed significantly by storage. We expect these findings to become of importance for the handling of purified stem cells used in clinical studies.
Our results show that the loss in cell viability was significant after 72 h. As total cell number decreased, part of the dying cells may have become decomposed so that less cell-sized entities could be detected; free cellular components, including DNA and enzymes, may thus be present in the stored cell product. Release of lactic acid has been described to correlate with lower viability in stored cells, especially for high cell concentrations. However, cell density was considerably lower in the CD133+ cell suspension we employed than in BM or comparable cell products (2,17). The apoptosis rate of freshly isolated CD133+ stem cells may seem considerable. Annexin V detects early apoptosis as opposed to the later stage of DNA fragmentation; in situ end labeling for DNA fragmentation detected between 2% and 3% apoptotic cells in immunomagnetic preparations from healthy donor s BM (19). Aprikyan et al., on the other hand, assessed 18% of freshly magnetic-activated cell sorting-purified CD34+ cells from BM as apoptotic and report a tripling of apoptosis rate by short-time cell culture (3).
Loss of metabolic activity preceded all other changes in stored CD133+ cells, occurring already after 30 h of storage (Fig. 5A) when colony-forming potential was still intact (Fig. 3). At the same time, a reduction of cell number was detected (Fig. 2C). Lower cell numbers may account for the lowered metabolic activity, though colony-forming potential remained unaffected. The decrease in proliferation deduced from CFU loss after 72 h, in turn, may have resulted from either cell death or a loss in stemness. However, we still detected substantial percentages of living cells coexpressing CD133 and CXCR4 after 72 h. qRT-PCR results displayed ongoing transcription of stemness-related genes as well. Pluripotency markers Lin-28 homolog A (LIN28A) and zinc finger protein 42 homolog (ZFP42, Rex-1) were expressed, as well as genes involved in the regulatory network of octamer-binding transcription factor 4 (Oct4), sex-determining region Y box 2(Sox2), and Nanog (PIPOX) (18) or involved in lineage decisions (REST, MED17) (9,11,42). The master pluripotency regulator Lin28 influences differentiation, as well as survival, and is important for HSC self-renewal (4,10,31,45,48). ZFP42 increased differentiation efficiency in mesenchymal stem cells (6,36). KRT18, an intermediate filament cytokeratin, has been described as an early differentiation marker in the embryo, whereas its zebrafish homolog is activated early in the regeneration process (8,30). Fibroblast growth factor 2 (FGF2), a proangiogenic chemokine secreted by different progenitor cell populations, has been shown to prevent differentiation of embryonic stem cells (22,25,33,49). The role these genes play in CD133+ HSC function is not yet clear; however, their ongoing expression is in accordance with the preserved proliferative capacity of CD133+ cells after 30 h.
High viability of stem cells in cell products for therapy is an important aim in order to secure the most effective treatment possible. Still, in relation to the high numbers of neutrophils infiltrating into ischemic cardiac tissue during reperfusion and undergoing apoptosis in situ, the impact of a limited number of damaged or dead transplanted cells may be considered marginal.
BMMNCs used for clinical trials display higher viability after 1 day of storage than freshly isolated CD133+ stem cells (35). During immunomagnetic selection, dead cells may be coenriched nonspecifically, and cell viability may be impacted by the purification process. Furthermore, in a stored BMMNC product, decomposing cells may be taken up by neighbor phagocytes, thus preventing the release of toxic cellular content and further cell damage.
In clinical multicenter trials with a core facility for cell preparation, whole BM needs to be transported before the specific cell population can be isolated. Lasky et al. confirm that in unpurified BM, cell viability, and colony-forming potential are well preserved for up to 3 days even at RT (20). Mean loss of viable CD34+ stem cells in BM was shown to remain below 10% after storage for 24 h, whereas mobilized stem cell products were much more sensitive to progenitor cell decrease over time (2). It may be assumed that short-time liquid storage of whole BM allows for successful purification of high-quality cell products, which may, in turn, be safely stored overnight before application.
To date, both short-term liquid storage and cryopreservation of purified HSCs have successfully been employed in clinical trials (14,43). In one case of relapsed leukemia with severe fungal infection, transplantation of isolated and cryopreserved CD133+ progenitor cells led to hematopoietic recovery (5). Adequate viability and function of stem cells derived from cryopreserved whole BM (21), cord blood (7), or peripheral blood progenitor cells (32) have been demonstrated as well. CD34+ cells may be separated from frozen and thawed BMMNCs with high efficiency and viability compared to cells isolated from fresh BM (32). However, cell loss may occur due to freezing and thawing procedures; Sartor et al. reported a mean viable CD34+ cell count reduction of 20% in cryopreserved BM samples (32). Above all, the common practice of cryopreservation, although allowing for long-term storage, carries a considerable risk of side effects. Commonly used cryoprotectants include dimethyl sulfoxide (26), which has been reported to cause severe side effects in patients after transplantation (16,46). Among the reported negative events, those of a cardiovascular nature stand out (12,41). Thus, liquid short-term storage should be preferred for cell products intended for cardiac transplantation.
To sum up our findings, we conclude that storage of BM-derived CD133+ stem cells for up to 30 h does not influence proliferation, expression of stemness markers, and endothelial-like differentiation capacity. However, we recommend keeping storage times as short as possible, as cell loss may occur. Well-designed point-of-care devices that allow for good manufacturing practice-conform cell processing in the operating theater may considerably improve feasibility and efficacy of stem cell transplantation in the future.
Footnotes
Acknowledgments
This work was supported by a “Verbundvorhaben” grant (Mecklenburg-Vorpommern, FKZ V630-F-075-2010/183) and by the Bundesministerium für Bildung und Forschung (BMBF; FK 0312138A). The authors thank Dr. Mike Essl from Miltenyi Biotec for very helpful discussion as well as Mrs. Margit Fritsche, Department of Cardiac Surgery, for excellent technical assistance. Contributions: Cornelia A. Lux and Peter Mark have equal shares in experiment conduction; Peter Mark contributed most of the assay and method design; Cornelia A. Lux undertook major parts of data analysis and writing. Christian Klopsch, Hoang Tu-Rapp, Wenzhong Li, Nan Ma, Gustav Steinhoff, and Robert David provided scientific advice and discussion to the present project, which was initiated and guided by G. Steinhoff. Michael Laupheimer contributed to the experimental work and data discussion. The authors declare no conflicts of interest.