
Abstract
Cartilage tissue engineering holds great promise for treating cartilaginous pathologies including degenerative disorders and traumatic injuries. Effective cartilage regeneration requires an optimal combination of biomaterial scaffolds, chondrogenic seed cells, and biofactors. Obtaining sufficient chondrocytes remains a major challenge due to the limited proliferative capability of primary chondrocytes. Here we investigate if reversibly immortalized mouse articular chondrocytes (iMACs) acquire long-term proliferative capability while retaining the chondrogenic phenotype. Primary mouse articular chondrocytes (MACs) can be efficiently immortalized with a retroviral vector-expressing SV40 large T antigen flanked with Cre/loxP sites. iMACs exhibit long-term proliferation in culture, although the immortalization phenotype can be reversed by Cre recombinase. iMACs express the chondrocyte markers Col2a1 and aggrecan and produce chondroid matrix in micromass culture. iMACs form subcutaneous cartilaginous masses in athymic mice. Histologic analysis and chondroid matrix staining demonstrate that iMACs can survive, proliferate, and produce chondroid matrix. The chondrogenic growth factor BMP2 promotes iMACs to produce more mature chondroid matrix resembling mature articular cartilage. Taken together, our results demonstrate that iMACs acquire long-term proliferative capability without losing the intrinsic chondrogenic features of MACs. Thus, iMACs provide a valuable cellular platform to optimize biomaterial scaffolds for cartilage regeneration, to identify biofactors that promote the proliferation and differentiation of chondrogenic progenitors, and to elucidate the molecular mechanisms underlying chondrogenesis.
Keywords
Introduction
Cartilaginous pathologies, ranging from degenerative disorders (osteoarthritis) to traumatic injuries, present formidable clinical challenges secondary to a lack of vascularization and limited regenerative capabilities (11,60). Articular cartilage damage occurs in acute trauma or through chronic degeneration, leading to pain and interference with normal activities of daily living. Surgical intervention, such as microfracture or arthroscopic debridement, can relieve associated pain but fails to regenerate normal tissue, instead producing fibrocartilage with inferior mechanical properties compared to healthy hyaline cartilage (56). Alternative therapies include autologous chondrocyte implantation, matrix autologous chondrocyte implantation, and matrix-associated autologous chondrocyte transplantation, procedures in which a small amount of articular cartilage is harvested from non-weight-bearing areas and expanded in vitro before implantation within a chondral defect (2,10,14,58). Implantation of a sufficient number of autologous chondrocytes into the defect may regenerate cartilage tissue with minimal immunologic response. Although promising, significant complications exist, including donor site morbidity, a limited yield of harvested chondrocytes, dedifferentiation during ex vivo expansion, and the need for two-stage operations (14,34). Moreover, proliferative capacity and postoperative tissue quality depends on the health of the joint, the age and health status of the donor, and the techniques of harvesting and culturing chondrocytes (1,18,44).
Chondrocytes are specialized cells derived from mesenchymal stem cells (MSCs). MSCs can differentiate along osteogenic, adipogenic, myogenic, or chondrogenic pathways depending on signaling pathways, transcription factors, cell-to-cell interactions, and many other factors (32,39). Thus, chondrogenic progenitors stimulated to differentiate along the chondrogenic pathway should be of therapeutic value (14). Mature articular chondrocytes produce, maintain, and reside within the cartilaginous extracellular matrix, consisting mainly of the large proteoglycan aggrecan and type II collagen, molecules that confer tensile strength and flexibility essential at the articular surface (1).
Cartilage regeneration has met significant challenges. Successful cartilage tissue engineering consists of at least three components: 1) biomaterial scaffolds that provide three-dimensional architecture resembling cartilaginous matrix, 2) chondrogenic cells or progenitors that can proliferate and differentiate into mature chondrocytes, and 3) potential biological factors that can facilitate the proliferation and differentiation of chondrogenic cells to overcome various cartilaginous pathologies (60). Primary chondrocytes have limited proliferative capability in two-dimensional culture (16—18,43). Although numerous attempts have been made to generate mature chondrocytes from embryonic stem cells, MSCs, or induced pluripotent stem cells (iPSCs), the outcomes have been mixed (57,59,60), largely because the molecular mechanisms underlying chondrogenesis from progenitor cells are not fully understood.
Here we investigate if reversibly immortalized mouse articular chondrocytes (iMACs) acquire long-term proliferative capability while retaining the chondrogenic phenotype. We immortalized primary mouse articular chondrocytes (MACs) with a retroviral vector expressing the SV40 large T antigen flanked with Cre/loxP sites. The iMACs proliferated in culture long term, although their proliferative capability can be effectively reversed by Cre recombinase. iMACs express chondrocyte markers and produce chondroid matrix in micromass culture, and when iMACs are implanted subcutaneously into athymic mice, cartilaginous masses are formed. Furthermore, the chondrogenic growth factor bone morphogenetic protein 2 (BMP2) promotes iMACs to produce more mature chondroid matrix resembling healthy articular cartilage. Taken together, our results demonstrate that iMACs acquire long-term proliferative capability without losing the intrinsic chondrogenic features of MACs. These findings should contribute significantly to the field of cartilage tissue engineering on several fronts. The long-term proliferative ability of iMACs will allow us to test and/or optimize biomaterial scaffolds for cartilage regeneration. iMACs can also be used to identify biofactors that promote the proliferation and differentiation of chondrogenic progenitors. Furthermore, iMACs provide a cellular platform for dissecting the molecular mechanisms underlying chondrogenesis.
Materials and Methods
Cell Culture and Chemicals
HEK-293 and C3H/10T1/2 cells were purchased from ATCC (Manassas, VA, USA). HEK-293 cells are human embryonic kidney cells transformed by human adenovirus E1a and commonly used as a recombinant adenovirus packaging line. C3H/10T1/2 cells are the established mouse embryonic fibroblasts that exhibit mesenchymal progenitor properties. Cells were maintained in complete Dulbecco's modified Eagle's medium (DMEM; Hyclone, Logan, UT, USA) at 37°C (3,9,19,20,29,32,45,49,54,67,68). The mouse embryonic fibroblast (iMEF) cells were isolated from the E12.5~E13.5 embryos of CD1 mice, reversibly immortalized with SV40 T antigen, and characterized as previously reported (27). Recombinant human interleukin-1β (IL-1β) was obtained from PeproTech (Rocky Hill, NJ, USA). All chemicals were purchased from Fisher Scientific (Pittsburgh, PA, USA) or Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted.
Construction of Recombinant Adenoviruses
Recombinant adenoviruses were generated using the AdEasy system (Agilent Technologies, Santa Clara, CA, USA) (9,12,23,24,32,36). The coding regions of human BMP2 and Cre recombinase were PCR amplified and cloned into an adenoviral shuttle vector to generate recombinant adenoviruses. These recombinant adenoviruses were used to transfect HEK-293 cells (21,36). The resulting adenoviruses were designated AdBMP2 and AdCre. The homemade adenovirus AdRFP was previously reported and used as a negative control (7,21,27,28,68). AdBMP2 and AdCre also express GFP (3,7,21,29).
Isolation of Primary Mouse Articular Chondrocytes (MACs)
All animal-related experiments in this study followed the animal use and care guidelines approved by the Institutional Animal Care and Use Committee of The University of Chicago. Primary MACs were isolated from the tibial plateau, femoral head, and femoral condyles of 5-day-old CD1 male mice [n = 5, parents purchased from the Harlan Laboratories (Indianapolis, IN, USA); bred at the University of Chicago Transgenic Animal Core Facility] and cultured as previously described (18,65). Isolated cells were plated in six-well plates (Corning Life Sciences, Corning, NY, USA) precoated with 0.1% gelatin solution. The pooled MACs within three passages were used for immortalization experiments.
Establishment of Reversibly Immortalized MACs (iMACs)
Prior to immortalization, the SSR #69 retrovirus was packaged by cotransfecting HEK-293 cells with SSR #69 and the packaging vector pCL-Ampho using LipofectAmine (Invitrogen, Carlsbad, CA, USA). Exponentially growing pooled primary MACs were infected with the packaged retrovirus SSR #69, which expresses SV40 large T antigen flanked with Cre/loxP sites (3,27—29,62,63). Cells were selected with hygromycin B and plated into 96-well plates (Corning Life Sciences) with serial dilutions. Single stable clones were chosen and expanded for further characterization. The resultant lines were designated as iMAC clones. AdCre infection was used to remove SV40 large T antigen for reversibility studies.
RNA Isolation and Semiquantitative RT-PCR (sqPCR) and Quantitative PCR (qPCR) Analyses
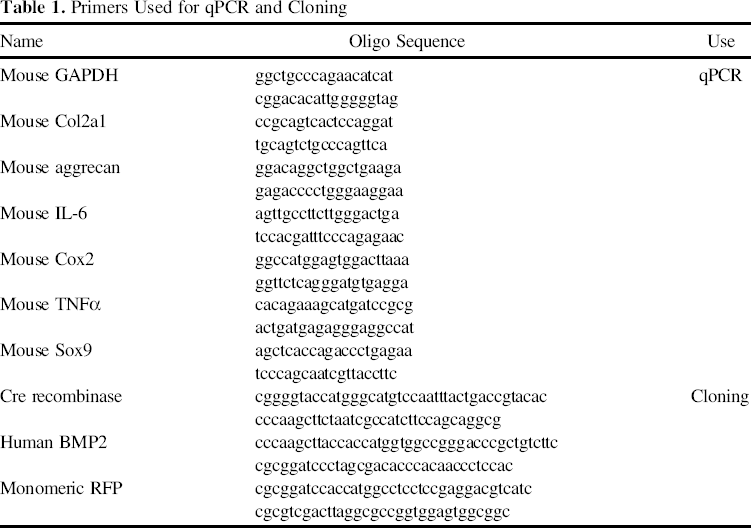
The sqPCR and qPCR analyses were performed as previously described (3,29,32,37,49,51,54,68). Briefly, total RNA was isolated from the iMAC clones, iMEFs, and C3H/10T1/2 cells using TRIzol reagents (Invitrogen). The cDNA products were generated for sqPCR analysis. Gene expression levels were normalized with GAPDH and amplified with mouse gene-specific primers (Table 1). PCR products were resolved and visualized on 1.2% agarose gels (Fisher Scientific). The prepared RT-PCR cDNA products were also used for quantitative real-time PCR (qPCR). Briefly, the qPCR primers were designed by using the Primer3 plus program to amplify mouse genes of interest. SYBR Green-based qPCR analysis (DyNAmo SYBR Green qPCR Kit; Fisher Scientific) was carried out using the Opticon DNA Engine 2 (Bio-Rad Laboratories, Hercules, CA, USA). The specificity of each qPCR reaction was verified by melting curve analysis and further confirmed by resolving the PCR products on 1.5% agarose gels (Fisher Scientific). Fivefold serially diluted pUC19 (New England Biolabs, Ipswich, MA, USA) was used as a standard. Triplicate reactions were carried out for each sample. Relative expression ratio was calculated by dividing the expression values of the gene of interest with GAPDH expression values.
Primers Used for qPCR and Cloning
Micromass Culture
Cells were grown in T75 flasks (Corning Life Sciences), collected, and resuspended in high density (2.5 × 109/ml). A total of 50 μl of the resuspended cell mix was carefully added to each well's center of 24-well cell culture plates (Corning Life Sciences) and allowed to attach for 4 h before adding complete DMEM. After 24 h, the media was changed to the StemPro Chondrogenesis Differentiation medium according to the manufacturer's protocol (Invitrogen).
Alcian Blue Staining
Alcian blue (Fisher Scientific) staining was performed on micromass culture as previously described (32,33). Briefly, iMACs, iMEFs, and C3H/10T1/2 cells were cultured in micromass as described above. Staining was performed at days 2, 3, and 4. The stain was extracted in 6 M guanidine hydrochloride (Fisher Scientific) overnight at room temperature, and the optical density was measured at OD540.
Immunohistochemical Staining
Col2aI immunohistochemical staining was performed on micromass culture formed by iMAC and MSCs. Micromasses were stained with rat anti-collagen type II antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by incubation with anti-rat IgG-HRP secondary antibody (Santa Cruz Biotechnology). The positive stain was visualized by DAB (Sigma-Aldrich) development. Staining without primary antibody and with rat IgG were used as negative controls.
Western Blot Analysis
Western blot analysis was performed as previously described (26,46,51). Briefly, subconfluent iMAC-5 cells were infected with AdCre (Cre) or AdRFP (RFP) for 48 h and subjected to Western blotting using an anti-large T antigen (at 1 μg/ml) (Santa Cruz Biotechnology). The total cellular proteins were normalized by the β-actin level using anti-β-actin antibody (Santa Cruz Biotechnology). HRP-conjugated secondary antibodies were from Pierce (Rockford, IL, USA). The proteins of interest were detected by using SuperSignal West Pico Chemiluminescent Substrate kit (Pierce).
Crystal Violet Staining
Crystal violet (Sigma-Aldrich) proliferation assays were utilized to assess cell viability and growth of iMACs in the presence of Cre as previously described (7,21,65). Briefly, iMAC clones were plated in 24-well plates at subconfluent conditions. Four hours after plating, cells were infected with AdCre or AdRFP. Cells were maintained for 3 to 5 days and then stained with crystal violet. The stain was dissolved in 10% acetic acid (Fisher Scientific), and optical density was measured at OD595.
Subcutaneous Injection of iMACs
All animal experimental procedures were approved by the IACUC. Athymic nude mice (male, 4—6 weeks old; Harlan Laboratories) were anesthetized using a mixture of ketamine (Henry Schein Animal Health, Dublin, OH, USA) and xylazine (Lloyd Laboratories, Shenandoah, IA, USA). When the mice were sufficiently anesthetized, 3.0 × 106 iMACs (infected with AdRFP or AdBMP2) were subcutaneously injected into the flanks. After 3 weeks, the mice were sacrificed, and masses at the injection sites were harvested with surrounding soft tissue. Retrieved masses were fixed with 10% formalin (Fisher Scientific), embedded in paraffin and stained with H&E (Fisher Scientific) or Alcian blue (Sigma-Aldrich) to assess for histology and chondroid matrix production.
Xenogen Bioluminescence Imaging
For bioluminescence imaging, mice were injected intramuscularly with firefly luciferase-tagged iMAC cells. At the indicated time points, animals were anesthetized with isoflurane (Baxter, Deerfield, IL, USA) attached to a nosecone mask (Xenogen, Alameda, CA) within Xenogen IVIS 200 imaging system (Xenogen) (8,21,22,35,37,38, 48,53). Mice were injected (IP) with D-luciferin sodium salt (Gold BioTechnology, St. Louis, MO, USA) at 100 mg/kg in 0.1 ml PBS. The pseudoimages were obtained by superimposing the emitted light over the grayscale photographs of the mice. Analysis was done with Xenogen's Living Image software.
Statistical Analysis
Microsoft Excel (Redmond, WA, USA) was used to calculate standard deviation (SD) and statistically significant differences between samples using the two-tailed Student's t-test. For all quantitative assays, each assay condition was performed in triplicate. A value of p < 0.05 was considered statistically significant when one comparison was being made. A Bonferroni correction was made when multiple t-tests were performed simultaneously to adjust the p value appropriately by dividing the critical p value (0.05) by the number of comparisons made (4).
Results
Primary Mouse Articular Chondrocytes (MACs) Are Effectively Immortalized Using SV40 Large T Antigen and Express Chondrocyte Markers
In this study, we isolated MACs from the tibial plateau, femoral head, and femoral condyles of 5-day-old CD1 mice (Fig. 1A, a and b). The isolated primary MACs were able to proliferate in culture, albeit slowly, and the morphology of primary MACs is shown at days 5 and 10 (Fig. 1A, c and d). To obtain immortalized and long-term cell lines, we utilized the retroviral vector that expresses SV40 large T antigen flanked with Cre/loxP sites (Fig. 1B) (62). We previously used this vector system to successfully obtain reversibly immortalized MEFs (iMEFs), fetal hepatic progenitors, and melanoblasts (3,27—29,63). We obtained more than 100 clones of iMACs that were derived from single immortalized cells through serial dilution cloning. iMACs after multiple passages seemingly still adapted a similar morphological phenotype to that of the primary MACs (Fig. 1C, a vs. b), indicating that the immortalization process did not significantly change the morphological phenotypes of primary MACs.

Isolation and immortalization of MACs. (A) Primary MACs were isolated and cultured from mouse tibial plateau, femoral head, and femoral condyles (a, b) as previously described (18,65). Primary chondrocytes are cultured and shown at 5 and 10 days after plating (c, d). (B) Schematic representation of the SSR #69 immortalization vector. The retroviral vector expresses the SV40 large T antigen, which is flanked with Cre/loxP sites and confers hygromycin B resistance. MACs were infected with Cre/loxP-flanked SV40 Large-T antigen retroviral vector. (C) Morphological comparison of primary and iMACs. MACs were infected with packaged SSR #69 retrovirus and selected against hygromycin. The resultant iMACs were obtained by serial dilution cloning. Subconfluent primary MACs (passage 2) (a) and iMACs (b) were photographed. Representative images are shown.
We evaluated the chondrocyte phenotype of the iMAC clones by first analyzing the endogenous expression level of collagen type II a 1 (Col2a1). Col2a1 encodes the proa-1 (II) chain of type II collagen, which is primarily found in cartilage. Using semiquantitative RT-PCR (sqPCR), we found that Col2a1 expression was readily detectable in all eight tested iMAC clones, although clones 1, 2, and 7 exhibited a lower expression level (Fig. 2A). As aggrecan is the most abundant proteoglycan in articular cartilage, we further analyzed its endogenous expression level in iMACs in comparison with that of the mesenchymal progenitor cells iMEFs and C3H/10T1/2 cells. Our sqPCR results indicated that both chondrocyte markers Col2a1 and aggrecan were highly expressed in the four iMAC clones, while their expression was barely detectable in iMEF and C3H/10T1/2 cells under the same amplification condition (Fig. 2B). The high levels of Col2a1, aggrecan, and sex-determining region Y-box 9 (Sox9) expression was also confirmed by quantitative real-time PCR analysis (Fig. 2C). As cytokines play an important role in the pathogenesis of osteoarthritis and/or other cartilage disorders, we examined the effect of rhIL-1b on the expression of tumor necrosis factor-α (TNF-α), IL-6, and cyclooxygenase 2 (COX2) in iMAC-5 cells. As shown in Figure 2D, rhIL-1β stimulation led to the elevated expression of TNF-α, IL-6, and COX2 in iMAC-5 cells. Taken together, these results indicate that the iMACs retain chondrogenic features.

Chondrocyte marker expression elevated in iMACs, but low in mesenchymal progenitor cells. Total RNA was isolated from representative eight clones of iMACs or the mesenchymal progenitor cells iMEF and C3H/10T1/2 cells. Semiquantitative RT-PCR (sqPCR) was performed using primers specific for the indicated mouse genes. (A) Endogenous Col2a1 expression in individual iMAC clones. The cDNA samples from eight representative iMAC clones were subjected to sqPCR analysis of Col2a1 expression. The samples were normalized with GAPDH expression level. (B) Expression status of Col2a1 and aggrecan in iMAC clones and mesenchymal progenitor cells. The cDNA samples from four iMAC clones, iMEFs, and C3H/10T1/2 cells were normalized with GAPDH and subjected to sqPCR analysis for the expression of Col2a1 and aggrecan. All assays were performed in at least three independent batches of experiments. Representative results are shown. (C). Endogenous expression of chondrocyte markers in iMACs and MSC lines. Total RNA was isolated from iMAC5, iMAC8, iMEFs, and C3H/10T1/2 cells and subjected to reverse transcription. The RT-PCR products were used for qPCR (SYBR Green-based, DAN Engine 2; BioRad) using primers specific for mouse Col2a1, aggrecan, Sox9, and GAPDH. Each assay condition was done in triplicate. Relative expression levels were calculated by dividing expression values of Col2a1, aggrecan, or Sox9 with that of GAPDH's. (D). Effect of IL-1β on IL-6, COX2, and TNF-α expression in iMACs. Subconfluent iMAC5 cells were cultured with serum-starved medium (DMEM supplemented with 1% FBS) for 12 h and then stimulated with rhIL-1β (10 ng/ml) for 2 h (PeproTech, Rocky Hill, NJ). Total RNA was isolated and subjected to RT. RT-PCR products were used for qPCR (SYBR Green-based, DAN Engine 2; BioRad) using primers specific for mouse IL-6, COX2, TNF-α, and GAPDH. Each assay condition was done in triplicate. Relative expression levels were calculated by dividing expression values of IL-6, COX2, and TNF-α by that of GAPDH.
iMACs Can Effectively Produce Chondroid Matrix in Micromass Culture
We next performed in vitro micromass culture in order to determine the ability of iMACs to produce chondroid matrix. Cell micromasses were formed by seeding highly concentrated iMACs, as well as the control MSCs (C3H/10T1/2 and iMEFs). Alcian blue staining qualitatively and quantitatively demonstrated significantly increased amounts of chondroid matrix production in iMAC clones (p < 0.025), when compared with MSC control groups (Fig. 3A). We also examined the Col2a1 expression in the micromasses by immunohistochemical staining and found that the two tested iMAC clones exhibited higher levels of Col2a1 expression than the C3H/10T1/2 cells (Fig. 3B). Thus, the cell micromass culture results further confirm that iMACs retain the chondrogenic phenotype.

Chondroid matrix production by iMACs in micromass assay. (A) Alcian blue staining of the micromasses formed by iMACs and mesenchymal progenitors. High cell density micromasses derived from iMAC-5, iMEF, or C3H/10T1/2 cells were seeded in multiwell culture plates. Alcian blue staining was performed at 2, 3, and 4 days following plating (a). The stain was then dissolved and quantified for OD595. iMACs demonstrate significantly higher chondroid matrix production (n = 3, p < 0.025). (B) Col2aI immunohistochemical staining was performed on micromass culture. Micromasses formed by the indicated cells were fixed and subjected to anti-collagen type II antibody immunohistochemical staining. Isotype IgG was used as a negative control staining (now shown). Each assay condition was done in triplicate. Representative results are shown.
Removal of Large T Antigen From iMACs Significantly Reduces Cell Survival and Proliferation
We tested if the immortalization phenotype of iMACs could be reversed by introducing the Cre recombinase. To efficiently deliver Cre recombinase into iMACs, we used a recombinant adenovirus that expresses the Cre recombinase (3,27—29,63). The iMACs were effectively infected by the adenoviral vectors (Fig. 4A). When the transduced iMAC cells were lysed and subjected to Western blotting analysis, the expression of SV40 T antigen was nearly undetectable in the AdCre-transduced cells (Fig. 4B). Furthermore, we found that Cre-mediated removal of the SV40 large T antigen significantly reduced the survival and proliferation of the affected iMACs as assessed by crystal violet staining qualitatively (Fig. 4C, a) or quantitatively (p < 0.01) (Fig. 4C, b). We further tested the potential tumorigenicity of the iMACs by performing intramuscular injections of firefly luciferase-labeled iMACs into athymic nude mice. By monitoring cell proliferation over time using Xenogen imaging, we found that the luciferase signal was undetectable for almost all mice within 2 weeks, and no masses were ever formed for up to 8 weeks (Fig. 4D). Thus, our results strongly suggest that the immortalization phenotype of the iMACs may be effectively reversed by the introduction of Cre recombinase and are not tumorigenic.

The Cre recombinase-mediated removal of SV40 large T antigen decreases the proliferative activity of iMACs. (A) Effective transduction of iMACs by recombinant adenoviral vectors. Subconfluent iMAC-5 cells were infected with AdCre (Cre) or AdRFP (RFP) for 36 h, and fluorescence signals were assessed. (B) Western blotting analysis of the large T expression in the iMACs transduced with Ad-Cre. Subconfluent iMAC-5 cells were infected with AdCre (Cre) or AdRFP (RFP) for 48 h and subjected to Western blotting using an anti-large T antigen. The total cellular proteins were normalized by the β-actin level. (C) Cell proliferation and survival assessed by crystal violet staining. Subconfluent iMACs were infected with AdCre or AdRFP for 3 or 5 days, fixed, and stained with crystal violet (a). The stain was dissolved and quantitatively measured for absorbance at 540 nm. The staining experiments were done in triplicate. Representative results are shown. Cell viability difference in iMACs infected with AdCre versus AdRFP was statistically significant (n = 3, p < 0.01). (D) In vivo growth of the iMACs cells in athymic nude mice. The iMAC5 cells were stably tagged with a retroviral vector expressing firefly luciferease, resulting in iMAC5-FLuc. Approximately 106 cells were intramuscularly injected into athymic nude mice (n = 5, male, 5—6 weeks old; Harlan Research Laboratories). At the indicated time points, the mice were subjected to bioluminescence imaging using Xenogen IVIS 200. The injection sites are indicated by the arrows. Representative results are shown.
iMACs Retain Chondrogenic Phenotype and Form Cartilaginous Masses In Vivo
Lastly, we investigated if iMACs would retain chondrogenic features in vivo. iMACs were first transduced with AdRFP or AdBMP2 and harvested for subcutaneous injection in athymic nude mice (Fig. 5A, a and b). Masses formed at the injection sites were retrieved after 3 weeks (Fig. 5A, c). While all mice injected with AdBMP2-transduced iMACs formed subcutaneous masses, only 6 of the 10 mice injected with AdRFP-infected iMACs formed small subcutaneous masses. H&E staining of the retrieved masses revealed that iMACs exhibited high proliferative activity when compared with normal articular cartilage (Fig. 5B, a). Furthermore, Alcian blue staining indicated that chondroid matrix production was readily detected in both iMAC clones. Furthermore, BMP2-transduced iMAC-5 cells seemingly produced more mature chondroid matrix, which resembles that of the healthy articular cartilage (Fig. 5B, b). It is noteworthy that the cartilaginous masses formed from iMACs usually reached peak sizes at week 3 postinjection and that no tumor-like masses were obtained for up to 8 weeks (data not shown). Taken together, these in vivo results strongly suggest that the immortalization manipulation of primary MACs may exert little effect on the chondrogenic features of iMACs.

Subcutaneous implantation of iMACs leads to formation of cartilaginous masses and chondroid matrix production, which can be potentiated by BMP2. (A) Subconfluent iMACs were infected with AdBMP2 or AdRFP, collected, and injected subcutaneously in athymic nude mice (n = 4 per group). Masses at the injection site (indicated in dotted yellow line) were harvested after 3 weeks (a, b). Subcutaneous masses were small, but grossly visible. The retrieved masses were homogenous in appearance and firm, but compressible to the touch (c). (B) The retrieved masses were paraffin embedded, sectioned, and subjected to H&E stain (a) and Alcian blue stain (b). Sections of healthy knee joints and femurs of athymic nude mice were used as a positive staining control. Representative results are shown.
Discussion
Successful cartilage repair should reduce pain, improve symptoms and long-term function, and prevent early osteoarthritis and subsequent total joint replacements by ultimately rebuilding hyaline cartilage instead of fibrous tissue (13,59,60). Current surgical treatments achieve less than optimal clinical outcomes. Thus, there is a critical need to develop new strategies in order to improve current clinical outcomes. One such strategy is to fabricate cartilage-like tissues to replace and/or repair the injuries and defects. Cartilage tissue engineering requires a combination of scaffolding, cells, signaling molecules, and biofactors to induce chondrogenesis. Scaffolds serve to provide mechanical function. Cells with a mature chondrocyte phenotype are required to synthesize and assemble extracellular matrix. Biofactors are needed to enhance cell differentiation, improve phenotypic activity, and recruit repair cells to injury sites.
One of the major challenges in cartilage tissue engineering is to obtain or expand sufficient seed cells that are able to function as mature chondrocytes. Although autologous chondrocytes (especially from hyaline cartilage) are considered the logical cells of choice, these cells are unable to grow well in monolayer culture and result in low cell yield from donor tissue (13,42,55,59,60). In this study, we demonstrate that reversibly immortalized iMACs acquire long-term proliferative capability while retaining the chondrogenic phenotype. Furthermore, the immortalization phenotype of iMACs can be effectively reversed by the introduction of Cre recombinase. We further demonstrate that the iMACs express chondrocyte markers and are able to produce chondroid matrix both in vitro and in vivo. The chondrogenic growth factor BMP2 promotes iMACs to produce more mature chondroid matrix resembling healthy articular cartilage. Thus, our results have demonstrated that iMACs acquire long-term proliferative capability without losing the intrinsic chondrogenic features of the MACs.
The reported findings should contribute significantly to the field of cartilage tissue engineering on several aspects. First, the long-term proliferative ability of iMACs will allow us to test and/or optimize biomaterial scaffolds for cartilage regeneration. Second, iMACs can be used to screen for and/or identify biological factors that promote the proliferation and differentiation of chondrogenic progenitors. Third, iMACs provide a reliable cellular platform for dissecting the molecular mechanisms underlying chondrogenesis. Furthermore, the reversible feature of the immortalization process offers an opportunity to validate any findings in a primary MAC-like condition.
It is noteworthy that numerous cell sources have been explored as potential seed cells for cartilage regeneration (13,14,42,55,57,59,60). MSCs have been induced to differentiate along the chondrogenic pathway using BMPs (61,64). BMP2 has been demonstrated as among the most chondrogenic growth factors, promoting chondrocyte differentiation in vitro and augmenting articular cartilage healing in vivo (6,30,40,50,52). Bone marrow-derived mesenchymal stem cells were shown to produce hyaline-like cartilage tissue (66). In recent years, adult MSCs derived from adipose tissues, satellite cells from muscle tissues, dermal fibroblasts, joint synovium, and human umbilical cord have been studied and shown varied degrees of chondrogenic potential (57,59,60). It seems that mesenchymal stromal cell-derived progenitors may be a better source for fibrocartilage tissue engineering rather than hyaline cartilage (13,60). It is conceivable that embryonic stem cells and iPSCs may also be used as potential chondrogenic progenitors. However, there is a significant gap in knowledge about how the stem cells and/or progenitor cells can be effectively directed along the chondrogenic lineage, and there is a need to elucidate these pathways.
We have successfully used the same strategy to generate several types of reversibly immortalized cells, including iMEFs, mouse fetal hepatic progenitor cells, and mouse melanoblastic cells (3,27—29,63). In each case, we found that, while the large T antigen enables the cells to achieve infinite proliferative activity, the immortalization process itself exerts little effect on the differentiation potential of these cells. This is consistent with these reported results about iMACs. The large T antigen encoded by SV40 plays essential roles in the infection of permissive cells and in the infection of nonpermissive cells (5,47). Primary MACs are nonpermissive for SV40, and the ability of SV40 large T antigen to immortalize MACs is largely dependent on its ability to complex with p53 (69). Thus, SV40 T antigen has become one of the most commonly used genes to immortalize primary mammalian cells. Other genes have been used to immortalize primary cells. The commonly used oncogenes include Kirsten ras (k-ras), c-myc, cyclin-dependent kinase 4 (CDK4), cyclin D1, B lymphoma Mo-MLV insertion region 1 (Bmi-1), human papillomavirus 16 (HPV 16) E6/E7, and telomerase (TERT), while the frequently inactivated tumor-suppressor genes are p53, Rb, and p16INK. Unlike cells immortalized by these oncogenes, SV40 large T antigen-transformed cells are, in general, not tumorigenic (15,25,31,41). Although the data was not shown, iMACs were not tumorigenic at least within the duration of 8 weeks in vivo.
In conclusion, we have demonstrated that reversibly iMACs acquire long-term proliferative capability while retaining the chondrogenic phenotype. The immortalization phenotype of iMACs can be effectively reversed by the introduction of Cre recombinase. We further demonstrate that the iMACs express chondrocyte markers and are able to produce cartilaginous matrix both in vitro and in vivo. The chondrogenic growth factor BMP2 promotes iMACs to produce more mature chondroid matrix resembling healthy articular cartilage. Thus, our results have demonstrated that the iMACs acquire long-term proliferative capability without losing the intrinsic chondrogenic features of the MACs. These findings should provide a valuable cell source for optimizing biomaterial scaffolds for cartilage regeneration, identifying biofactors that promote the proliferation and differentiation of chondrogenic progenitors, and elucidating the molecular mechanisms underlying chondrogenesis.
Footnotes
Acknowledgments
The authors thank Dr. Philippe Leboulch of Harvard Medical School for providing the SSR #69 vector. The reported work was supported in part by research grants from the National Institutes of Health (R.C.H., H.H.L., and T.C.H.), North American Spine Society (T.C.H.), and the SHOCK Fund (S.H.H.). J.D.L. was the recipient of the Pritzker Research Fellowship from The University of Chicago Pritzker School of Medicine and the Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship. This work was also supported in part by The University of Chicago Core Facility Subsidy grant from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health through Grant Number UL1 TR000430. The authors declare no conflicts of interest.