
Abstract
Radiotherapy is an effective teatment for brain tumors but often results in cognitive deficits in survivors. Transplantation of embryonic or brain-derived neural stem/progenitor cells (BNSPCs) ameliorated cognitive impairment after irradiation (IR) in animal models. However, such an approach in patients requires a clinically relevant source of cells. We show for the first time the utilization of enteric neural stem/progenitor cells (ENSPCs) from the postnatal intestinal wall as a source of autologous cells for brain repair after injury caused by IR. Cells were isolated from the intestinal wall and propagated in vitro for 1 week. Differentiation assays showed that ENSPCs are multipotent and generated neurons, astrocytes, and myofibroblasts. To investigate whether ENSPCs can be used in vivo, postnatal day 9 mice were subjected to a single moderate irradiation dose (6 or 8 Gy). Twelve days later, mice received an intrahippocampal injection of syngeneic ENSPCs. Four weeks after transplantation, 0.5% and 1% of grafted ENSPCs were detected in the dentate gyrus of sham and irradiated animals, respectively, and only 0.1% was detected after 16 weeks. Grafted ENSPCs remained undifferentiated but failed to restore IR-induced loss of BNSPCs and the subsequent impaired growth of the dentate gyrus. We observed microglia activation, astogliosis, and loss of granule neurons associated with grafted ENSPC clusters. Transplantation of ENSPCs did not ameliorate IR-induced impaired learning and memory. In summary, while autologous ENSPC grafting to the brain worked technically, even in the absence of immunosuppression, the protocols need to be modified to improve survival and integration.
Introduction
Radiotherapy remains an essential and efficient treatment tool for malignant brain tumors. However, cranial irradiation (IR) often results in debilitating intellectual and cognitive deficits, such as impairment of learning and memory. These deficits have been shown to be more severe after cranial radiotherapy in younger children and are often called late effects since they develop over time (12,19,52). The underlying pathogenesis of IR-induced injury is not well understood, but it has been closely linked to loss of postnatal neurogenesis (34). Neurogenesis is widely accepted to occur throughout life in discrete regions of the brain, the subventricular zone and the dentate gyrus (DG) of the hippocampus (4,5,15,28). These two regions harbor neural stem/progenitor cells continuously generating new neurons, which have been shown to play a role in maintaining learning and memory (57). Owing to their high proliferative capacity, these cells are susceptible to IR damage (16,35,42). IR-induced depletion of neural stem/progenitor cells appears to be long lasting, even after a single moderate dose of radiation (8,24). Interventions after IR have been investigated. Voluntary physical exercise was shown to increase the number of hippocampal neural stem/progenitor cells and the rate of neurogenesis after IR of the young mouse brain and partially normalized the IR-induced behavior alterations (38). Cellular intervention after IR is still not extensively studied and limited to a few reports. Grafting of human embryonic and neural stem cells into the hippocampus of irradiated adult immune-deficient animals ameliorated IR-induced cognitive impairments (1,2). Considering the importance of the recipient's immune response and inflammation on migration, cell survival, and differentiation of grafted cells, we have recently shown that grafting of syngeneic neural stem cells in immune-competent animals generates neurons and astrocytes in irradiated brains (48). However, such an approach requires a clinically relevant source for neural stem cells. The enteric neural stem/progenitor cells (ENSPCs) exist in the enteric nervous system (ENS) and have been proposed as a valuable source for autologous cell therapy for the central nervous system (CNS) (50,53). ENSPCs develop from the neural crest during embryogenesis (47,58) and reside in the adult intestinal wall in two separate locations, the myenteric plexus and the submucosal plexus in rodents and humans (27,44). ENSPCs can easily be accessed using laparoscopy and obtained as mucosal biopsies or in the form of intestinal pieces, such as the vermiform appendix (32,50).
In the current study, we grafted syngeneic ENSPCs into the hippocampus of control and irradiated young mouse brains. We evaluated the survival and differentiation of the grafted cells, the host tissue immune response, and the outcomes of grafting of ENSPCs on the endogenous stem cell pool and memory function of the animals after IR.
Materials and Methods
Animals
Female C57/BL6 mice (Charles River, Sulzfeld, Germany) were used for the entire experiment. Animals were housed in equal light/dark cycles (12/12 h) and given access to food and water ad libitum. All experimental procedures were conducted in accordance with European and Swedish animal welfare regulations and approved by Gothenburg and Stockholm ethical committees (applications No. 361/11and N9/12).
Isolation and Culture of Enteric Neural Stem/Progenitor Cells
Cells were isolated from postnatal day (P) 10–15 mouse pups. Animals were anesthetized with isoflurane (Isobavet; Schering-Plough Corporation, Kenilworth, NJ, USA) and decapitated. The whole body was sprayed with 70% ethanol, and a midline incision was made to expose the abdominal cavity. The intestine was removed and placed in ice-cold phosphate-buffered saline (PBS) supplied with penicillin/streptomycin (100 U penicillin and 100 μg streptomycin/ml; Gibco/Invitrogen, San Diego, CA, USA). In a sterile laminar flow hood, intestines were cut into approximately 2-cm-long pieces, and the muscosal and submucosal layers were gently scraped off using fine 45° angled forceps. The remaining muscular layers containing the myenteric plexus were placed into transfer medium HibernateA (Gibco/Invitrogen). Tissue pieces were minced with a scalpel blade and incubated for 40 min at 37°C in a digestion solution consisting of 0.01% papain (Worthington/Cell Systems, Lakewood, NJ, USA), 0.1% dispase II (Roche, Mannheim, Germany), 0.01% DNase (Worthington/Cell Systems), collagenase IV (Worthington/Cell Systems), 12.4 mM MgSO4 (Sigma-Aldrich, St. Louis, MO, USA) in Hank's buffered salt solution (HBSS) without Ca2+ and Mg2+ (Gibco/Invitrogen), followed by gentle trituration using a 1-ml pipette. The enzymatic digestion was stopped by adding the culture medium Neurobasal-A (Gibco/Invitrogen) supplemented with B27 (1:50), Glutamax (2 mM), and penicillin/streptomycin (100 U penicillin and 100 μg streptomycin/ml) (all from Gibco/Invitrogen). The cell suspension was passed through a 40-μm cell strainer (BD Biosciences, San Jose, CA, USA) and centrifuged at 300 × g for 5 min. After two washing steps in the culture medium, the cell pellet was resuspended in 1 ml culture medium, and the live cells were counted using the trypan blue (Gibco/Invitrogen) exclusion method. Cells were cultured nonadherently (neurosphere assay) at a density of 2 × 105/ml in fresh above-mentioned culture medium supplemented with 20 ng/ ml bFGF (PeproTech, London, UK), 20 ng/ml epidermal growth factor (EGF, Gibco/Invitrogen), 10 ng/ml glial cell-derived neurotrophic factor (GDNF, Gibco/ Invitrogen), and 2 μg/ml heparin (Sigma-Aldrich). Cells were cultured for 7 days at 37°C and 5% CO2. The culture medium was changed every other day.
Cell Characterization, Differentiation, and Immunocytochemistry
Cell characterization and/or differentiation were performed using either single cells or neurospheres. To produce single cell suspensions, neurospheres were collected by centrifugation (300 × g for 5 min). The cell pellet was resuspended in 0.5 ml TrypleE express (Gibco/Invitrogen) and incubated at 37°C for 2 × 3 min (with 15–20 times trituration in between). Fresh medium was then added, and the cell suspension centrifuged. The cell pellet was resuspended in 1 ml fresh medium for counting. For characterization, cells (seeded at 3 × 104 densities)/neurospheres were plated onto poly-L-ornithine (100 μg/ml; Sigma-Aldrich)-coated glass coverslips in culture medium with the growth factors and incubated for 2–3 h at 37°C and 5% CO2. For differentiation, cells/neurospheres were plated onto poly-D-lysine/laminin-coated eight-well culture slides (BD Biosciences). Cells were allowed to differentiate for 7–10 days at 37°C and 5% CO2 in culture medium without addition of the growth factors. For immunocytochemistry, cells/neurospheres were fixed with 4% paraformaldehyde (PFA; Sigma-Aldrich) for 20 min at room temperature. After several washes with Tris-buffered saline (TBS; Sigma-Aldrich), cells/neuropheres were blocked for 1 h with a solution containing 5% or 10% normal donkey serum (Jackson ImmunoResearch Lab, West Grove, PA, USA) for cells and neurospheres, respectively, 0.1% Triton X-100 (Sigma-Aldrich), TBS, and then incubated overnight with the primary antibodies (Table 1). After several washes, cells/neurospheres were then incubated with the secondary antibodies (Table 1) for 1 h at room temperature. ToPro3 (1:1,000; Molecular Probes/Invitrogen, Eugene, OR, USA) was used as a nuclear counterstain. The mounting medium ProLong Gold (Molecular Probes/ Invitrogen) was used for coverslipping.
List of Antibodies
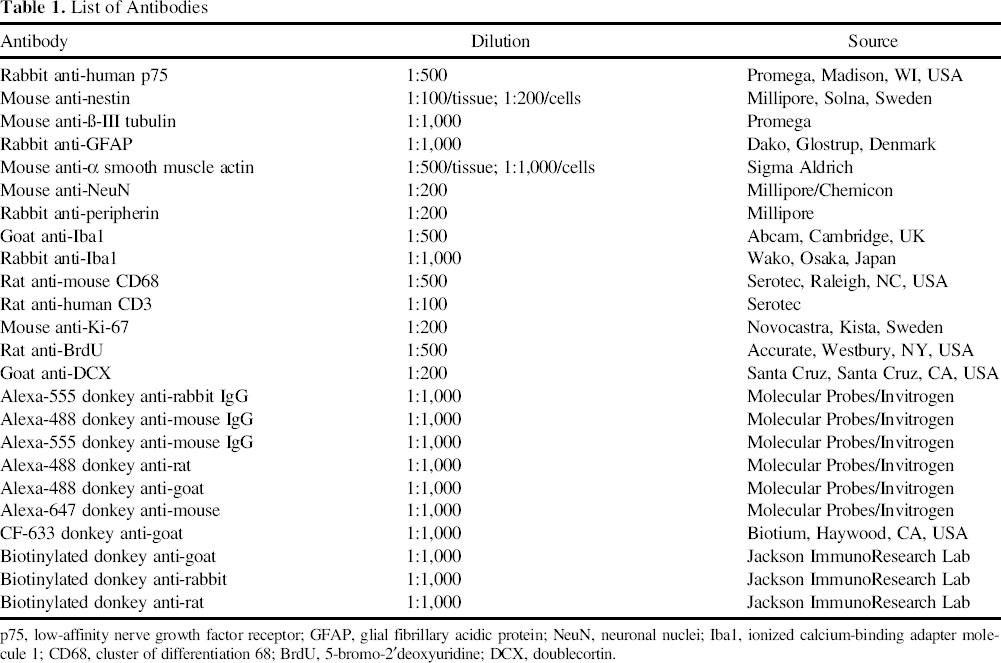
p75, low-affinity nerve growth factor receptor; GFAP, glial fibrillary acidic protein; NeuN, neuronal nuclei; Iba1, ionized calcium-binding adapter molecule 1; CD68, cluster of differentiation 68; BrdU, 5-bromo-2′deoxyuridine; DCX, doublecortin.
Irradiation
P9 mice were anesthetized with an intraperitoneal (IP) injection of 50 mg/kg tribromoethanol (Sigma, Stockholm, Sweden) and randomly divided into IR or sham-IR (SH; only anesthetized) groups. Animals were placed in prone position (head to gantry) on a polystyrene bed. The whole brain was irradiated using a linear accelerator (Varian Clinac 600CD, Radiation Oncology Systems, San Diego, CA, USA) with 4 MV nominal photon energy and a dose of 2.3 Gy/min. The irradiation source to skin distance was about 99.5 cm. The head was covered with 1 cm tissue equivalent material to obtain an even radiation dose throughout the underlying tissue. The dose variation within the target volume was estimated to be ±5%. A single dose of either 6 or 8 Gy was administered (Fig. 1A, B), and then animals were returned to their biological dams for recovery.

In vivo experimental design. (A) The first experimental setup. Animals were subjected to 8 Gy IR on postnatal day 9 (P9), received intrahippocampal injections of cells (C) in one hemisphere and vehicle (V) in the other hemisphere on P21, and were perfused 4 weeks after engraftment. (B) The second experimental setup, where animals were subjected to a moderate irradiation dose (6 Gy) on P9 and bilateral intrahippocampal injections of either vehicle or cells on P21. Animals were assigned to three experimental groups: sham irradiated injected with vehicle (SH + V), irradiated injected with vehicle (IR + V), or irradiated injected with cells (IR + C). Learning behavior was evaluated 8 weeks after the grafting using the IntelliCage platform. BrdU was administered on 3 consecutive days prior to the animals being perfused 16 weeks after transplantation.
Cell Labeling and Grafting Procedure
On the grafting day, neurospheres were split, and a single cell suspension was generated as mentioned above. Cells were labeled with the lipophilic dye CM-DiI (referred to as DiI for the rest of the text; Invitrogen). Briefly, 2 × 106 cells were incubated in a solution consisting of 2 μg DiI per 1 ml culture medium (Neurobasal-A with growth factors) at 37°C for 5 min followed by incubation at 4°C for 15 min. Cells were washed twice with the culture medium and resuspended in fresh culture medium (with growth factors) at a final concentration of 5 × 104/μl. Mice (P21) were initially anesthetized with 5% isoflurane and maintained with 1.5% during surgery. Animals received 2 μl intrahippocampal injections of either cell suspension or vehicle (culture medium with growth factors) using a 10-μl Exmire microinjection syringe (Ito Corporation, Tokyo, Japan) at the following coordinates relative to bregma anterior/posterior: −1.8 mm, lateral: ±1.7 mm, ventral: −2.9 mm. The injection was done over 1 min, and the syringe remained in the injection site for 5 min to reduce the backflow of the injecta and slowly withdrawn thereafter. Two experimental setups were designed (Fig. 1A, B). In the first setup, which was intended to evaluate the survival and differentiation of grafted ENSPCs, IR (n = 4) or SH (n = 4) animals received cells in one hemisphere and the vehicle in the other hemisphere and were perfused 4 weeks later. In the second setup, which was intended to evaluate the effect of grafted ENSPCs on the animal's learning behavior and to assess the endogenous neural stem cell pool after IR, animals were divided into three groups: SH injected with vehicle (SH + V; n = 15), irradiated injected with vehicle (IR + V; n = 15), or irradiated injected with cells (IR + C; n = 15), and they received bilateral intrahippocampal injections of either cell suspension or vehicle. After finishing the transplantation procedure, the remainder of labeled cells was allowed to differentiate in vitro as mentioned above to ensure that the labeling process did not affect their differentiation capacity.
Administration of 5-Bromo-2′Deoxyuridine
To evaluate proliferation and survival of the endogenous neural stem/progenitor cells after ENSPC transplantation, animals received single daily IP injections of 5-bromo-2′deoxyuridine (BrdU; Sigma-Aldrich) for 3 consecutive days prior to perfusion (Fig. 1B).
Tissue Preparation
Animals were deeply anesthetized with sodium pentobarbital, transcardially perfused with 0.9% sodium chloride (Sigma-Aldrich), and then fixed with 4% PFA in 0.1 M phosphate buffer (pH 7.4). Brains were collected and postfixed in the same fixative for 24 h, then transferred to 30% sucrose (Sigma-Aldrich) in 0.1 M phosphate buffer for cryoprotection, and left for a minimum of 3 days. Brains were cryosectioned sagittally using a sliding microtome (Leica SM2000R, Wetzlar, Germany) into 25-μm-thick free-floating sections and stored as 1:12 series at 4°C in tubes containing cryoprotection solution (25% glycerin, 25% ethylene glycol in 0.1 M phosphate buffer; all from Sigma-Aldrich) for further histological analysis.
Immunohistochemistry and Immunofluorescence
Sections were incubated in sodium citrate (Sigma-Aldrich) pH 6.0 for 20 min at 80°C when antigen retrieval was needed. When the immunoperoxidase method was used, the endogenous peroxidase was quenched by incubating sections in 0.6% H2O2 (Sigma-Aldrich) for 30 min at room temperature. The nonspecific binding was blocked by incubating sections in a solution of 3% normal donkey serum (Jackson ImmunoResearch Lab) and 0.1% Triton X-100 in TBS for 1 h at room temperature. Sections were incubated at 4°C for 24–72 h with the primary antibodies (Table 1), followed by 1-h or 2-h incubation with biotinylated or fluorescent secondary antibodies, respectively (Table 1). ToPro3 (1:1,000) was used as a nuclear counterstain when fluorescence staining was used. For visualization of the immunoperoxidase stain, sections were incubated for 1 h with avidin–biotin solution (1:100; Vectastain ABC Elite kit, Vector Laboratories, Burlingame, CA, USA). The stain was developed with H2O2, nickel chloride, and 3–3′diaminobenzidine tetrahydrochloride (DAB; 1:100; Saveen Werner AB, Malmö, Sweden). Sections were mounted onto glass slides and coverslipped using NeoClear® and NeouMount® (Merck, Whitehouse Station, NJ, USA) for immunoperoxidase staining or Pro-Long Gold antifade for the fluorescent staining.
Microscopy and Histological Analysis
Characterization and differentiation of the in vitro grown cells was assessed using a confocal laser-scanning microscope (TCS SP2; Leica). The histological analysis for the in vivo experiments was performed in all sections containing the dorsal hippocampus. To evaluate the survival of the transplanted ENSPCs, DiI-labeled cells (DiI+) were quantified in the DG in six series section intervals. Stacks of images for the entire structure were acquired in the confocal microscope using the 20× objective lens and digital zoom of 3. Cells were considered for counting when DiI staining surrounded at least two thirds of the nucleus (i.e., ToPro3 stained). The total number of cells that survived was the sum of all counted cells multiplied by the series interval. To evaluate the differentiation of transplanted cells, 100 DiI+/ToPro3+ cells were quantified and examined for coexpression of the suggested phenotypic marker for each cell type. The expression of cluster of differentiation 68 (CD68) and CD3 was also evaluated using the confocal microscopy. All confocal microscopy analysis was performed using a pinhole setting of 1.0 airy with sequential fluorochrome excitation scans at 488 nm, 546 nm, and 633 nm. The analysis of microglia was performed by quantifying ionized calcium-binding adapter molecule 1-positive (Iba1+) cells in the entire DG in two to three sections per animal, spaced 300 μm apart. BrdU-positive (BrdU+) and doublecortin-positive (DCX+) cells were counted in the subgranular zone (SGZ) and the granule cell layer (GCL), respectively, in five to six sections per animal, spaced 150 μm apart. All quantification was done using the stereology software (Stereo Investigator, MicroBrightField Inc., Williston, VT, USA). The total number of cells was the sum of all counted cells multiplied by the series interval. The cell density per structure was determined as the total number of quantified cells divided by counting volume.
IntelliCage
The IntelliCage (NewBehavior, Zurich, Switzerland) platform gives us the possibility to examine animal learning behavior in a home cage environment over a period of time with minimal handling (26,60). One week prior to starting the experiment, animals were briefly anesthetized with isoflurane and subcutaneously implanted with microtransponders (Planet ID GmbH, Essen, Germany) to allow detection of each animal. Animals were accommodated in IntelliCages 8 weeks after the ENSPC grafting. The experiment was performed using the protocol described previously (26). Briefly, after a 5-day introduction period, each animal was randomly allocated to one corner during three testing periods, where the corner was changed every 5 days. Care was taken to not assign one animal to a corner more than once. Animals were returned to their regular cages immediately after the last day of the experiment. The analysis was only performed during the active period (dark), and visits lasting longer than 3 min or visits where no nose poke occurred were not considered.
Statistical Analysis
All values were presented as mean ± SEM. An unpaired Student's t-test was used when comparing two groups. A one-way ANOVA was used when comparing more than two groups. A two-way ANOVA was used to analyze the microglia number at 4 weeks after grafting. IntelliCage data were analyzed using repeated-measures two-way ANOVA. Bonferroni post hoc tests were applied in all analyses. All statistical analyses were performed using the software GraphPad Prism 5 (San Diego, CA, USA). Significance was considered when p < 0.05.
Results
In Vitro Characterization and Differentiation
Cells isolated from the muscular layers/myenteric plexus were grown as nonadherent with growth factors for 7 days. After 2 days in culture, cell aggregates were visible, and on day 7, neurospheres were well established (Fig. 2A). The cells were immune reactive for low-affinity nerve growth factor receptor (p75) and nestin (Fig. 2B, C), indicating that they were ENSPCs. Following this culture protocol, we were able to expand cells once. After being under differentiation conditions (withdrawal of growth factors) for 7–10 days, ENSPCs were capable of differentiating into neurons (bIII tubulin-positive; bIII+), astrocytes (glial fibrillary acidic protein-positive; GFAP+), and myofibroblasts (a smooth muscle actin-positive; aSMA+) (Fig. 2D–F). Moreover, since we prelabeled cells with the lipophilic dye CM-DiI before grafting to track them in vivo, labeled cells were also differentiated in vitro and were bIII+, GFAP+, and aSMA+ (Fig. 2G–I).

In vitro characterization and differentiation. (A) ENSPC neurospheres on day 7. Scale bar: 200 μm. (B and C) Cells expressed low-affinity nerve growth factor receptor (p75; red) and nestin (green) when stained as neurospheres (B) or single cells (C); scale bar: 25 and 50 μm (B and C, respectively). (D–F) Cells were able to differentiate into βIII-tubulin+ cells (D), glial fibrillary acidic protein-positive (GFAP+) cells (E), and α smooth muscle actin (αSMA+) cells (F) after withdrawal of growth factors and being in culture for 7–10 days. Scale bar: 100 μm (D) and 50 μm in (E and F). (G–I) Cells were able to differentiate in vitro after being labeled with the lipophilic dye CM-DiI (DiI; red) before grafting, and generate βIII-tubulin+ cells (green) (G), GFAP+ cells (green) (H), and αSMA+ cells (green) (I). Scale bar: 25 μm in (G and H) and 50 μm in (I). ToPro3 (blue) were used for nuclear staining (B–I).
ENSPCs Remain Undifferentiated When Grafted Into the Brain
To investigate whether the ENSPCs can survive and differentiate in the brain, ENSPCs were intrahippocampally transplanted into healthy (SH) or injured (IR) brains of young mice (P21). Four weeks after transplantation, 0.5% (mean ± SEM = 576 ± 16) and 1% (mean ± SEM = 1,194 ± 15) of the grafted ENSPCs (DiI+) were detected in the DG of the SH (SH + C) and IR animals (IR + C), respectively (Fig. 3A), and no DiI+ cells were seen in the vehicle-injected hemisphere (not shown). The majority of grafted cells were found within the dorsal part of the GCL forming large clusters (Fig. 3B); however, ENSPCs were also detected in the ventral part of the GCL, the hilus, and the molecular layer (Fig. 3C). Further, we were interested in examining whether the ENSPCs can differentiate in the brain. All DiI+ cells were found to be expressing p75 and nestin (p75+/nestin+), and no colocalization of DiI+ cells with neuronal nuclei (NeuN; mature neuronal marker), peripherin (peripheral neuronal marker), GFAP (astrocyte), or α-SMA (myofibroblast) was seen, indicating that the cells remained undifferentiated (Fig. 3D–I). Furthermore, we did not see colocalization of DCX (immature neuronal marker) in DiI+ cells (Fig. 3J), suggesting that the grafted cells were not even under delayed differentiation. Further, we asked whether the grafted cells are still proliferating since they remained undifferentiated and colonize in large clusters; however, no DiI/Ki-67 (proliferation marker) colocalization was seen (Fig. 3K).

Characterization of grafted cells 4 weeks after transplantation. (A) Bar graph shows number of grafted ENSPCs (DiI+) detected in the DG of the hemisphere injected with cells in sham irradiated (SH + C) and irradiated (IR + C) animals; n = 4; *p = 0.03. (B and C) Grafted cells (DiI+; red) in DG. DiI+ cells were mostly clustered within the dorsal blade of the GCL (B) but were sometimes detected within the GCL ventral blade, the hilus (h), and the molecular layer (m) (C). (D) All DiI+ cells (red) expressed p75 (green) and nestin (blue). Inset in (D) enlarged in (E). (F–J) No differentiation was observed in vivo. (F) Neuronal nuclei (NeuN; green), (G) peripherin (peri; green), (H) GFAP (green), (I) a smooth muscle actin (aSMA; green), and (J) doublecortin (DCX; green). (K) No expression of Ki-67 (green) was detected in DiI+ cells (red). ToPro3 (blue) was used for nuclear staining in (B, C, and F–K). Scale bar: 100 μm in (B, C, and J); 50 μm in (D); 30 μm in (F–I and K); 25 μm in (E).
Transplantation of ENSPCs Disrupts the GCL and Provokes Neuroinflammation
IR and SH animals received cells in one hemisphere and vehicle in the other hemisphere to examine the effects of the grafted ENSPCs within the same individual. Four weeks after transplantation, disruption of the GCL structure in the hemisphere injected with cells, but not the vehicle, was noticed in all animals. This was found to be either due to thinning of the GCL or nearly complete loss of GCL neurons in the layer where the grafted cells integrated (Fig. 4A, B). Further, we looked at the host tissue immune reaction. We found a significant effect of ENSPC transplantation on the number of microglia (Iba1+) (F = 7.21; p = 0.02). The number was significantly increased in SH + C animals compared to SH injected with vehicle (SH + V), but there was no difference in irradiated brains (Fig. 4C). Moreover, activated microglia (Iba1+/CD68+) were seen surrounding and infiltrating the ENSPC clusters (Fig. 4D). DiI-labeled fragments were frequently seen within Iba1+/CD68+ cells (Fig. 4E), suggesting the phagocytic activity of these microglia. Furthermore, we observed upregulation of hypertrophied astrocytes (GFAP+) in the hemisphere injected with cells, and all ENSPC clusters were surrounded by pronounced astrogliosis (Fig. 4F). No CD3+ cells (pan T-cell marker) were seen (Fig. 4G).

Tissue integrity and inflammation 4 weeks after grafting. (A) Images of the GCL 4 weeks after grafting. The GCL (visualized with ToPro, blue) remained intact (upper panel) in animals injected with vehicle (Veh), while GCL degeneration (lower panel) was seen in animals injected with cells (DiI; red). (B) A similar image as in the lower panel in (A) stained for NeuN (green), confirming the loss of the granule neurons. (C) Bar graph showing the number of microglia [ionized calcium-binding adapter molecule 1-positive (Iba1+) cells] in the DG of vehicle- or cell-injected hemispheres of sham and irradiated animals 4 weeks postgrafting. n = 4; effect of treatment: #p = 0.02; *p = 0.05. (D) Activated microglia Iba1 (blue)/cluster of differentiation 68 (CD68; green) surrounding and infiltrating ENSPC (DiI+; red) cluster in the dorsal blade of the GCL. (E) An activated microglial cell, Iba1 (blue)/CD68 (green), which has engulfed DiI-labeled fragments. (F) Reactive astrocytes (GFAP+ cells; green) surrounding the DiI+ cell cluster in the dorsal GCL. (G) No CD3+ cells (T-cells; green) were detected. ToPro3 was used as nuclear staining in (A, B, F, and G). Scale bar: 100 μm in (A, C, and G); 25 μm in (D and F); 20 μm in (E).
Grafting of ENSPCs Triggered Long-Term Microglia Accumulation, But Had No Effect on Hippocampal Neurogenesis
To investigate the long-term effects of ENSPCs, animals were perfused 16 weeks after transplantation. At this time point, we only detected 0.1% of grafted cells (DiI+) in the DG of IR + C animals (mean ± SEM; 127.5 ± 41.21). Next, we quantified Iba1+ cells in the DG. IR + C animals displayed a significant increase in Iba1+ cell numbers compared to IR + V (mean ± SEM; IR + V: 5,231 ± 343.7; IR + C: 6,713 ± 512.2; p < 0.02), but no difference in number compared to SH + V (mean ± SEM = 6,590 ± 231.5) (Fig. 5A). Since IR decreases the volume of the brain (18), particularly of the DG (17,46), we further proceeded to evaluate cell densities in all three groups to avoid any confounding effects of volume alterations. IR + C showed the highest Iba1+ density compared to SH + C and IR + V (mean ± SEM; SH + V: 13,121 ± 291; IR + V: 13,794 ± 283; IR + C: 17,200 ± 886; p < 0.001) (Fig. 5B). Last, we examined the impact of ENSPC transplantation on hippocampal neurogenesis. To assess the proliferation of endogenous neural stem cells, animals were injected with BrdU for 3 days prior to sacrifice (Fig. 1B), and we quantified BrdU+ cells in the SGZ. Irradiated, IR + V and IR + C, animals showed approximately 90% reduction in BrdU+ cells compared with SH + V (p < 0.001) (Fig. 5C), representing 75% and 80% reduction in BrdU+ cell density in IR + V and IR + C, respectively (p < 0.001) (Fig. 5D). When we quantified DCX+ cells to assess endogenous neurogenesis, we also found about 90% reduction in numbers of DCX+ cells in the GCL of IR + V and IR + C compared to SH + V (Fig. 5E), representing 80% and 85% reduction in DCX+ cell density in IR + V and IR + C, respectively (p < 0.001) (Fig. 5F).

ENSPCs triggered long-term inflammation and did not restore hippocampal neurogenesis. Bar graphs showing the effects of ENSPC grafting 16 weeks after transplantation; sham irradiated injected with vehicle (SH + V), irradiated injected with vehicle (IR + V), and irradiated injected with cells (IR + C), n = 8 in each group. (A and B) The number and density of microglia (Iba1+ cells) in the DG, respectively. *p = 0.02; ***p < 0.001. (C and D) The number and density of BrdU+ cells, respectively. ***p < 0.001. (E and F) The number and density of DCX+ cells, respectively. ***p < 0.001. ns, not significant.
Grafting of ENSPCs Impaired Learning in Young Irradiated Mice
We were interested in investigating the effect of ENSPC transplantation on the animals' learning. Animals were divided into three groups, SH injected with vehicle (SH + V), IR injected with vehicle (IR + V), and IR injected with cells (IR + C). We assessed the learning behavior using the IntelliCage platform 8 weeks after transplantation. We found that IR + C animals had the highest error ratios (incorrect events/total events) regarding the visits to the incorrect corners, nose pokes to nonallocated corners, for the first and second corners, and spent more time during the incorrect visits (Fig. 6A–C). No clear difference in these parameters was seen during the last training period, corner 3 (data not shown); however, during this period, IR + C animals scored the highest ratio in visiting the previously allocated corner (corner 2) (Fig. 6D), indicating that reversal learning was also impaired.

Assessment of learning 16 weeks after grafting. Learning was assessed using the IntelliCage for 3 weeks. Sham irradiated injected with vehicle (SH + V), irradiated injected with vehicle (IR + V), and irradiated injected with cells (IR + C), n = 15 in each group. IR + C showed impaired learning. (A) The ratio of incorrect visits during corner 1 and 2 periods. (B) The ratio of incorrect nose pokes during corner 1 and 2 periods. (C) The time spent during the incorrect visits. #Interaction between the treatment and time: p = 0.04. (D) Nose poke ratio in the previously correct corner during the corner 2 and 3 periods as an assessment for reversal learning. IR + C animals scored the highest error ratios during the training period of corner 3, indicating impaired reversal learning ability. *Effect of treatment: p = 0.04.
Discussion
Stem cell therapy is a promising therapeutic approach for numerous CNS disorders and injuries. Very few reports have addressed the topic of intervention after brain IR (1,2). The recipient's immune response governs the survival, migration, and differentiation of grafted cells (7,21), so we decided to graft syngeneic cells to immune-competent animals, as in our recent study of BNSPCs (48). Such an approach requires a clinically relevant source of neural stem cells. Induced pluripotent stem cells (iPSCs) are expected to be able to generate a variety of cell types derived from the same patient (54). However, this process is time consuming, associated with problems of improper reprogramming, and rejection, even if the donor is genetically identical (40,59). Harvesting neural stem cells from the brain is, in most cases, too invasive and is not likely a fruitful approach. Against this background, we decided to investigate the feasibility of grafting the more easily accessible ENSPCs into the brain.
ENSPCs exist in the ENS and have been suggested as a source for autologous cell therapy for CNS disorders (50,53). ENSPCs are identified by their expression of the low-affinity nerve growth factor receptor (p75) and nestin (3,53). ENSPCs can produce neurons, astrocytes, and myofibroblasts in vitro (6,49), as well as when transplanted into intestines (20,41). We were able to isolate p75+/nestin+ cells from the muscular layer/myenteric plexus, and the isolated ENSPCs were able to generate neural cells (neurons and astrocytes) and myofibroblasts when allowed to differentiate in vitro (Fig. 2A–F). Notably, we were able to expand the isolated ENSPCs only once, suggesting that ENSPCs are more restricted progenitors and have limited self-renewal capacity, at least under the culture conditions we applied. To track the grafts in vivo, ENSPCs were labeled with the lipophilic dye CM-DiI. Labeled cells were also able to differentiate in vitro to neural cells and myofibroblasts (Fig. 2G–I), suggesting that the labeling procedure did not affect their differentiation capacity.
To test if they survive in the brain, we grafted syngeneic ENSPCs into the hippocampus of irradiated and nonirradiated young mice (Fig. 1A, B). Similar to the BNSPCs (2,48), the majority of the grafted ENSPCs were found within the GCL in both normal and irradiated animals, suggesting a homing mechanism for stem cells in this region, the nature of which remains to be elucidated. One month after transplantation, we found 1% of the grafted cells in the DG of the irradiated animals and only half of this amount in the nonirradiated animals (Fig. 3A). This stands in contrast to our results after grafting of BNSPCs, where we found approximately 6% of the grafted cells in the GCL, with no difference between nonirradiated and irradiated brains (48). The labeling techniques were different, though. The BNSPCs were labeled with BrdU, not DiI, and the use of BrdU has been questioned (11). However, assuming that labeling indicates that the cells, or their progeny, were grafted, survival was lower for ENSPCs than BNSPCs. Death of transplanted cells is a major hindrance for cell therapy (31), because of the small number of remaining cells to be incorporated, because of the local reaction caused by dying cells (55), and because of the structural changes incurred (48). Long-term studies have revealed a low yield of grafted cells over time (2,45). Likewise, 4 months after transplantation, the number of detectable ENSPCs was only 0.1%, indicating that death of grafted ENSPCs is an ongoing process.
After IR of the growing brain, the loss of endogenous NSPCs leads to a long-lasting, progressive reduction of neurogenesis (8), so a strategy aiming to replenish both undifferentiated NSPCs and differentiated granule neurons would appear ideal. Transplanted BNSPCs have the capacity to produce neural cells in different CNS disease models. In the adult brain of immune-competent animals, the chronic inflammation resulting from IR presumably prevented neuronal differentiation of grafted cells (35), whereas the transient inflammation in the young brain (23) did not interfere with neuronal differentiation if grafting was performed 1 week or later after IR (48). Our in vivo results demonstrated that ENSPCs were lacking the expected differential phenotypic markers 4 weeks after transplantation (Fig. 3F–J), and all grafted cells maintained their expression for the putative enteric stem/progenitor cell markers (p75 and nestin) in both irradiated and nonirradiated animals (Fig. 3D, E), suggesting that the lack of differentiation is not a result of the IR. Interestingly, when BNSPCs were cocultured in vitro with ENSPCs, or transplanted into different parts of the gastrointestinal tract, they were able to differentiate, and mostly adopted enteric neuron properties (13,29,33). Moreover, enteric neurons and glia were able to survive in the brain for long periods when the myenteric plexus was implanted into the hippocampus and striatum (30,56). Taken together, this might suggest that undifferentiated ENSPCs dominate, or fail to differentiate, because of a lack of the factors required for ENSPC differentiation in the brain. This is a possibility that needs further investigation. Not only cell origin but also culture history has been shown to affect integration and differentiation, for example, primary DG cells robustly generated neurons, whereas DG cells cultured for 7 days did not (10). Hence, further investigation of both cell origin and culture conditions is warranted.
Transplantation of cells into the brain is inevitably traumatic. In addition, the grafted cells may cause further morphological disruption, as demonstrated after grafting of syngeneic BNSPCs into irradiated and nonirradiated brains (48). In the present study, the loss of granule neurons was even larger. The GCL degeneration was clearly associated with the presence of grafted ENSPCs (Fig. 4A, B), signifying that the damage was not caused by the cell delivery procedure, but was rather cell related. The cause of the GCL loss is not clear, but it is possibly because of inflammation. Neuroinflammation (microgliosis and astrogliosis) results in neuronal loss due to secretion of proinflammatory cytokines and generation of free radicals (36). Microglia, the primary immune cells in the brain, are continuously surveying for unusual events (39). Clearance of cellular debris is one of the primary functions of microglia (37). In this context, hypoxia leads to acute cell death of the grafts, which consequently stimulates microglial response (45). In line with this, we observed numerous microglia after engraftment of ENSPCs, particularly in nonirradiated brains (Fig. 4C), and microglia near ENSPCs exhibited phagocytic activity as indicated by their expression of CD68 (43) (Fig. 4D). Four months after the grafting, accumulation of microglia was evident in the irradiated animals (Fig. 5A, B), despite the fact that IR to young brains normally only causes transient inflammation (23) and leads to loss of microglia (25). Along with microglial activation, we also observed pronounced astrocytic reactivity (astrogliosis) (Fig. 4F). Under lesion conditions, astrocytes become reactive and accumulate to form a barrier between the lesioned and the healthy tissue (51). Here, ENSPC clusters were entirely surrounded by astrocytes and appeared isolated from the rest of the brain tissue in a manner resembling the glial scarring after brain lesions (Fig. 4F). This feature of glial reactivity (both microglia and astroglia) was seen as a sign of rejection of mesenchymal allografts, even in the absence of T-lymphocyte involvement (11,55). Given that we used syngeneic ENSPCs, and no T-cells (CD3+ cells) were seen (Fig. 4G), this likely suggests that the ENSPCs were rejected through innate (resident) immune mechanisms. Collectively, the observed inflammatory reaction might be responsible for the GCL loss and, in addition, could also create a hostile milieu not permissive for ENSPC survival and differentiation.
Beneficial outcomes of transplanted cells have been demonstrated in diverse CNS disease models. Stem cells exert their beneficial effects either by replacement or secretion of trophic factors (paracrine effects) that modulate the inflammatory response or stimulate the endogenous stem cell pool, which eventually is reflected by positive behavioral outcomes (9,14,22). In our study, neither replacement nor inflammatory modulation is likely, since no in vivo differentiation was seen, and rather intense inflammation was morphologically linked to ENSPCs. Our data showed that ENPSC grafting could restore neither the proliferation of hippocampal neural stem cells (BrdU+ cells in SGZ) nor the hippocampal neurogenesis (DCX+ cells in the GCL) (Fig. 5C–F). Furthermore, we also observed that irradiated animals that received ENSPCs performed worse in the learning tests (scored the highest error ratios) (Fig. 6A–C) and displayed poor reversal learning (as judged by visits to the previously correct corner) (24) (Fig. 6D). This could be the result of the ongoing inflammatory reaction and subsequent loss of granule neurons. In summary, no beneficial effects of grafting could be demonstrated, at least not with the current culture and grafting protocols.
Conclusion
To our knowledge, we demonstrate here for the first time the effects of ENSPC transplantation into the young mouse hippocampus. ENSPCs remained undifferentiated, provoked neuroinflammation, caused a loss of the GCL, and impaired learning. However, despite the negative effects observed, we cannot completely rule out transplantation as a useful therapeutic approach for CNS repair, as the outcome might be different in other brain injuries and anatomical locations. Alternative culture conditions, modulation of the recipient's immune response (despite the use of syngeneic cells), or reprogramming of cells might be necessary to make the ENSPCs efficient and alter the outcome.
Footnotes
Acknowledgments
The authors would like to thank Rita Grandér for assistance with the irradiation procedure. This work was supported by the Swedish Childhood Cancer Foundation (Barncancerfonden), the Swedish Research Council (Vetenskapsrådet), governmental grants from the agreement concerning research and education of doctors (ALF) in Stockholm and Gothenburg, the Swedish Cancer Foundation (Cancerfonden), the Sten A. Olsson's Foundation, the King Gustav V Jubilee Clinic Research Foundation (JK-fonden), the Frimurare Barnhus Foundations of Gothenburg and Stockholm, the Wilhelm and Martina Lundgren Foundation, the Gothenburg Medical Society, the Aina Wallström and Mary-Ann Sjöblom Foundation, and the Ulla and Rune Amlöv Foundations. The authors declare no conflict of interest.