
Abstract
The success of pancreatic islet transplantation is limited by delayed engraftment and suboptimal function in the longer term. Endothelial progenitor cells (EPCs) represent a potential cellular therapy that may improve the engraftment of transplanted pancreatic islets. In addition, EPCs may directly affect the function of pancreatic β-cells. The objective of this study was to examine the ability of EPCs to enhance pancreatic islet transplantation in a murine syngeneic marginal mass transplant model and to examine the mechanisms through which this occurs. We found that cotransplanted EPCs improved the cure rate and initial glycemic control of transplanted islets. Gene expression data indicate that EPCs, or their soluble products, modulate the expression of the β-cell surface molecule connexin 36 and affect glucose-stimulated insulin release in vitro. In conclusion, EPCs are a promising candidate for improving outcomes in islet transplantation, and their mechanisms of action warrant further study.
Keywords
Introduction
The pancreatic islet is a complex, multicellular structure comprising multiple endocrine and nonendocrine cell types. In the native pancreas, islets are densely vascularized with specialized, fenestrated endothelium (2) and receive 10% of pancreatic blood flow despite only making up 1–2% of tissue mass (20). The vascular endothelium is crucial for pancreatic development (6,24), and the importance of vascular endothelial cells for the survival and proper function of mature insulin-producing β-cells is beginning to become apparent (15,21,24,32,42). The impact of the intraislet endothelium is likely to be twofold. Firstly, as a conduit for blood supply, intraislet vasculature is crucial for the delivery of oxygen, sensing of blood glucose, dissemination of insulin, and removal of waste products. Secondly, within the islet, endothelial cells are in intimate contact with β-cells and directly enhance insulin transcription and secretion and stimulate β-cell proliferation (21,42). This may occur through the provision of humoral factors, production of basement membrane components, or via cell contact-dependent mechanisms.
Pancreatic islet transplantation is a promising therapy for the treatment of type 1 diabetes (25), but the majority of β-cells die in the early postoperative period (13). Starting with the organ procurement stage, through islet isolation, transport and culture, and posttransplantation, islets face a myriad of cell stresses including hypoxia and immunemediated damage (16,19,41). Posttransplantation, islet revascularization can take up to 1 month, and the ultimate vascular density of the islet is often compromised. The provision of exogenous endothelial cells, or their precursors, at the time of transplantation is one strategy to improve islet revascularization and/or enhance β-cell function.
Endothelial progenitor cells (EPCs) are a circulating bone marrow-derived cell population first described by Asahara and colleagues (1). EPCs are able to home to sites of tissue damage or ischemia and can participate in wound healing, postnatal vasculogenesis, and reendothelialization of blood vessels (18,35). While EPCs have the capacity to integrate into newly growing vessels, probably more important is their provision of humoral factors at the tissue damage site, which may recruit accessory cells, support vessel remodeling, and elicit prosurvival responses.
This study examines the potential of EPCs to enhance the engraftment of transplanted pancreatic islets and investigates in vitro the mechanisms through which EPCs, or their secreted factors, modify islet cell gene expression and function.
Materials and Methods
Antibodies and Reagents
Recombinant mouse tumor necrosis factor-α (TNF-α) was obtained from R&D Systems (Minneapolis, MN, USA). The following antibodies were used for experiments: anti-mouse E-selectin antibody (clone UZ6; Abcam, Cambridge, UK; used at 10 μg/μl), immunoglobulin G1b (IgG1b) isotype control (Abcam; used at 10 μg/μl), anti-rat Alexa488 (Life Technologies, Mulgrave, Australia; used at 10 μg/μl), mouse fragment, crystallizable region (Fc) block (BD Biosciences, San Jose, CA, USA; used at 5 μg/μl), phycoerythrin (PE)-labeled antimouse cluster of differentiation 31 (CD31; Clone MEC 13.3; BD Biosciences; used at 1:50), PE-labeled isotype control (BD Biosciences; used at 10 μg/μl), guinea pig anti-insulin (Abcam; used at 1:3,200), rabbit antifluorescein (Life Technologies; used at 10 μg/μl), biotinylated donkey anti-guinea pig (Abacus, East Brisbane, Australia; used at 1:100), and goat anti-rabbit Alexa647 (Life Technologies; used at 10 μg/μl). Fluorescein-labeled tomato lectin was obtained from Vector Laboratories (Burlingame, CA, USA). Mefloquine was obtained from Sigma (St. Louis, MO, USA) and streptavidin- labeled cyanine 3 (Cy3) from Jackson Laboratories (West Grove, PA, USA; used at 1:100).
Endothelial Progenitor Cell Isolation and Culture
EPCs were derived from male C57B6 mice (Laboratory Animal Services, The University of Adelaide, Adelaide, Australia) as previously described (4,14). Bone marrow cells were obtained by flushing femurs and tibiae of 6- to 12-week-old mice using M199 medium (Sigma #M0393). Cells were cultured on fibronectin (50 μl/ml; Roche, #10838039001; Basel, Switzerland) in 20% fetal calf serum (Gibco/Invitrogen, Carlsbad, CA, USA). Medium was supplemented with endothelial cell growth supplement and heparin (both at 15 μg/ml; BD Biosciences, #356006; a tissue extract containing multiple growth factors including fibroblast growth factor, VEGF, and endothelial cell growth factors α and β), and heparin (BD Biosciences) (both at 15 μg/ml). Cells were harvested with 0.1% trypsin-ethylenediaminetetraacetic acid (EDTA) 2–3 min at 37°C (Sigma Aldrich, St. Louis, MO, USA) within 7 days of initial seeding. EPC-conditioned media was obtained from 90% confluent day 7 EPC cultures, centrifuged (800 × g/5 min), sterile filtered (0.4 μm), and used immediately. In some experiments, EPCs were labeled with carboxyfluorescein diacetate succinimidyl ester (CFDA; 10 μM; Vybrant Cell Tracer kit; Life Technologies) for 10 min at 37°C and washed thoroughly immediately prior to cotransplantation.
TNF-α Treatment of EPCs
EPCs were treated with TNF-α (5 ng/ml) for 5 h. Cells were harvested with 0.1% trypsin–EDTA (2–3 min at 37°C) and stained with primary antibodies for 30 min at 4°C. Cells were stained with secondary antibodies (30 min at 4°C) and analyzed with the flow cytometer (Beckman Coulter XL-MCL; Gladesville, Australia).
Murine Islet Isolation and Culture
All experimental procedures were approved by the Animal Ethics Committee of the University of Adelaide and conform to the guidelines established by the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes. Pancreatic islets were isolated from 6- to 12-week-old male C57B6 mice (University of Adelaide Laboratory Animal Services, SA, Australia). Briefly, 3 ml cold M199 medium (Sigma) containing 0.67 mg collagenase (Liberase TL grade; Roche) per pancreas was infused into the pancreatic duct in situ. The pancreas was removed and digested at 37°C for 14–16 min. Islets were purified on a discontinuous Ficoll gradient (GE Healthcare, Buckinghamshire, UK). Following extensive washing, islets were cultured free floating (37°C, 5% CO2) in Roswell Park Memorial Institute medium (RPMI; Sigma) supplemented with l-glutamine, penicillin, streptomycin, and 10% fetal calf serum (RPMI–FCS) for up to 4 days. For coculture experiments, 25 islets per well were handpicked to be plated on nonpermeable Transwell inserts (0.4-μm pores; Corning, Clayton, Australia) above EPC monolayers seeded onto fibronectincoated wells. For contact coculture experiments, islets (50 per well) were added to EPC monolayers and cultured for 4 h prior to RNA extraction (RNAeasy Mini Kit; Qiagen, Hilden, Germany).
Detection of Intraislet Endothelial Cells
Handpicked islets were dissociated with Accutase (37°C, 5 min; Sigma), strained through a tube-top strainer [BD Bioscience (BD Falcon), San Jose, CA, USA] and recovered in RPMI–FCS for 30 min at 4°C. Following blocking, cells were stained with directly conjugated antibodies for 30 min at 4°C and analyzed at the flow cytometer (Beckman Coulter XL-MCL; Gladesville, Australia) and using FCS express software (version 3.0, De Novo Software, Los Angeles, CA, USA).
Quantitative Real-Time PCR
Grafted kidneys were homogenized into RLT buffer using a TissueLyserII (Qiagen). RNA was isolated from kidney homogenates or purified islets using QIAshredder columns and an RNAeasy Mini Kit (Qiagen) and treated to remove genomic DNA (TURBO DNA-free kit; Ambion, Life Technologies), prior to reverse transcription (Omniscript; Qiagen). Real-time quantitative PCR analysis was performed on triplicate samples using the following gene-specific Taqman® primers (Applied Biosystems; Life Technologies): insulin 1 (INS; Mm01259683_g1), connexin 36 (Cx36, Mm00439121_m1), connexin 43 (Cx43; Mm00439105_m1), connexin 45 (Cx45; Mn01253027_m1), E-cadherin (Ecad) (Cdh1; Mm01247357_m1), vascular endothelial growth factor A (VEGFA; Mm01281449_m1), hepatocyte growth factor (HGF; Mm01135193), collagen type 1 a 1 (Col1A1; Mm00801666_g1), integrin b1 (ITGB1; Mm01253230_m1), and fibronectin (Fn1; Mm01256744_m1). β-2-microglobulin (B2M; Mm00437762_m1) was used as a housekeeping gene to normalize expression data, and hypoxanthine-guanine phosphoribosyltransferase (HPRT; Mm00446968_m1) was used as a confirmatory housekeeper. Mean normalized expression values were calculated using Qgene Module software (Kansas State University, Manhattan, KS, USA; http://www.qgene.org/qgene/index.php) as previously described (27).
Immunohistochemistry
The graft-bearing kidney was harvested, fixed in buffered formalin (10% buffered formalin, ACE Chemical Company, Camden Park, Australia), and processed into paraffin blocks. Serial sections (5 μm) were taken through the graft, and every 10th section was processed for hematoxylin–eosin (Sigma-Aldrich) staining to identify the graft site. Sections were dewaxed in xylene and rehydrated prior to antigen retrieval with Proteinase K (Roche Australia, Dee Why, Australia) at 37°C for 20 min. Sections were blocked with 10% normal donkey serum (Sigma-Aldrich) and processed for immunofluorescence detection of insulin and fluorescein (fluorescein-labeled tomato lectin). Digital images were obtained using an Olympus BX50 fluorescence microscope (Macquarie Park, NSW, Australia) with LED illumination. Images were prepared using ImageJ software (NIH, Bethesda, MD, USA; http://rsb.info.nih.gov/ij).
Intraperitoneal Glucose Tolerance Test
Mice were fasted for 18 h and given 2 g/kg glucose [d-(+)-glucose solution (10%); Sigma Aldrich] by intraperitoneal (IP) injection. Tail vein blood samples were taken prior to injection and at 15, 30, 60, and 120 min postinjection. Whole blood was analyzed for glucose using a glucometer (Optium, Abbott Diabetes Care, Victoria, Australia) and using the low range of a high-sensitivity murine insulin ELISA kit (Crystal Chem, Inc., Downers Grove, IL, USA).
Islet Transplantation Under the Kidney Capsule
C57B6 mice were rendered diabetic by IP injection of 180–200 mg/kg streptozotocin (Sigma-Aldrich) in citrate buffer (BDH Chemicals, Kilsyth, VIC, Australia). Diabetes was confirmed by two consecutive blood glucose readings higher than 16.6 mM (39). Diabetic mice were transplanted with a minimal mass (200 islets) of cultured islets under the kidney capsule with or without day 7 EPCs (1 × 106 cells). Cure of diabetes was defined as the first day of two consecutive nonfasted blood glucose readings of <11.1 mM with no subsequent reversion to hyperglycemia (two consecutive readings above 11.1 mM).
Glucose-Stimulated Insulin Release
Islets were handpicked into basal Krebs buffer (3 mM glucose; BDH Chemicals) at 37°C (basal). Groups of 10 islets were transferred into fresh basal Krebs buffer (3 mM glucose) at 37°C for 1 h. In some experiments, the Cx36 inhibitor mefloquine (10 μM) was included throughout the culture periods. Islets were spun briefly and supernatant replaced with Krebs buffer with high glucose (20 mM) at 37°C for 10 min (first phase). Islets were spun briefly, and the supernatant was replaced with Krebs buffer with high glucose (20 mM) at 37°C for 50 min (second phase). Following recovery in basal buffer for 1 h, islets were incubated with high potassium Krebs buffer (70 mM; High K+; BDH Chemicals) at 37°C for 30 min. Islets were spun, and supernatant was collected. Total pellet proteins were extracted by resuspending islets in RIPA buffer [0.1% sodium dodecyl sulfate (SDS); 100 mM phenylmethansulfanyl fluoride serine protease inhibitor; BDH Chemicals] and subjecting to 10 freeze–thaw cycles in liquid nitrogen. Protein concentration was estimated using the EZYQ Protein Estimation Assay (Life Technologies) as per the manufacturer's instructions. Supernatants were analyzed for insulin concentration using the Ultra Sensitive Mouse Insulin ELISA Kit (low-range assay; Crystal Chem, Inc.). Stimulation indices were calculated by dividing insulin secretion values at basal glucose levels by those detected in the first phase of high glucose.
Statistics
Endothelial cell enumeration data over time was compared by one-way ANOVA with Bonferroni posttest. Cure rates were compared between groups by logrank (Mantel–Cox) test. Blood glucose levels within the revascularization period (days 3–14 posttransplant) were analyzed using area-under-the-curve (AUC) analysis of log10 transformed data, and AUC values were compared by one-tailed unpaired t tests. Mean normalized gene expression data were compared by unpaired t tests. Analyses were performed using Prism 5 for Windows (Version 5.04; Graphpad, San Diego, CA, USA) software.
Results
Intraislet Endothelial Cells Are Rapidly Lost in Culture
To investigate the persistence of endothelial cells in murine pancreatic islets during culture, endothelial cells were enumerated in isolated islets by flow cytometry (Fig. 1). At various timepoints, handpicked islets were dispersed into single-cell suspensions, stained for the endothelial cell marker CD31, and analyzed by flow cytometry (Fig. 1A). In freshly isolated islets, CD31+ endothelial cells comprised 2.5 ± 0.1% of total islet cells. After 1 day of culture, 73 ± 20% of the endothelial cells persisted, and following 4 days of culture, this was significantly reduced to 12 ± 3% of the original number (p < 0.005) (Fig. 1B). These data indicate that following islet isolation, CD31+ intraislet endothelial cells are rapidly lost from the islet structure during culture.
Intraislet endothelial cell density decreases rapidly during islet culture. Isolated islets (25–60 islets per well) were dispersed and endothelial cells enumerated at different timepoints during culture by flow cytometric detection of cluster of differentiation 31 (CD31). (A) Representative flow cytometry dot plot. Dotted line represents gating region for CD31–PE+ cells within dispersed islets. (B) Endothelial cell density is expressed as mean ± SEM relative to the values at day 0. *p < 0.05 compared to day 4; **p < 0.005 compared to day 0; n = 4 mice. PE, phycoerythrin; FSC-A, forward scatter.
Cotransplanted Endothelial Progenitor Cells Speed the Engraftment of Transplanted Pancreatic Islets
To examine whether EPCs could enhance the engraftment of pancreatic islets, EPCs were cotransplanted under the kidney capsule of diabetic mice with a marginal mass of islets. Since we were interested in whether EPCs can replenish the endothelial cells that are lost during culture (Fig. 1), we used islets that had been cultured in vitro for 3 days prior to transplantation. To obtain EPCs, bone marrow cells were cultured with a commercial endothelial cell growth supplement (containing VEGF and fibroblast growth factor) on fibronectin-coated plates to promote the vascular endothelial lineage. Our extensive characterization of these cells has been previously published (4). Briefly, these EPCs were shown to express CD144 at their cell surface and were positive for CD34, c-kit, stem cell antigen-1 (sca-1), fetal liver kinase 1 (Flk-1; VEGFR2), CD133, CD146, CD31, endothelial nitric oxide synthase (eNOS), von Willebrand factor (vWF), Tunica interna endothelial cell kinase (Tie2; angiopoietin-1 receptor), chemokine C-X-C motif receptor 2 (CXCR2), CXCR4, α5-integrin, β1-integrin, CD45, and CD11b surface antigen, CD14 low and negative for CD105, and CD90 message (4). Moreover, they form capillary-like tubes in vitro using the three-dimensional Matrigel assay (4). Herein, we further validate their endothelial lineage (Fig. 2) by showing that they efficiently bound fluoresceinlabeled tomato lectin (which selectively binds rodent vascular endothelium; Fig. 2A) and upregulated E-selectin upon TNF-α treatment (Fig. 2B). In initial transplantation experiments, the marginal dose of day 3 cultured islets required to cure approximately half of recipients was determined as 200 islets. To examine the effect of EPC cotransplants, mice received this marginal mass of islets, with or without EPCs, under the kidney capsule and were monitored daily for nonfasted blood glucose and body weight. There was a significantly improved cure rate in cotransplanted animals compared to controls (p = 0.002) (Fig. 3). Revascularization in the mouse occurs within 14 days posttransplantation (21). Thus, following the expected revascularization period, the percentage cure in cotransplanted animals at day 14 was 83%, compared to 20% in the islets-only control group. There was significantly improved posttransplant weight gain in the mice receiving cotransplanted EPCs compared to mice receiving islets alone (data not shown; p = 0.007). Blood glucose levels during the revascularization period (days 3–14) in cotransplanted animals were significantly lower compared to the islets-alone group (p = 0.002) (Fig. 3D). To examine the ultimate in vivo function of transplanted islets among the groups, only cured animals were challenged with a bolus of IP glucose 28 days after transplantation (Fig. 3E). Owing to the small numbers of cured animals in the islets-only group, differences were not significant. Diabetic mice receiving EPCs only or sham operations did not experience improved glycemic control following transplant (Fig. 4), suggesting that EPCs act to enhance the function of transplanted islets rather than causing the regeneration of endogenous islets. Thus, cotransplantation of EPCs improved the cure rate and glycemic control of a marginal mass of syngeneic islets into STZ-treated diabetic mice. To examine the in vivo fate of cotransplanted EPCs, CFDA-labeled EPCs were cotransplanted with islets, and tissues were harvested at various timepoints posttransplant. EPCs were detected under the kidney capsule at day 7 in close proximity to the graft (Fig. 3F) but were undetectable at day 28. EPCs were absent from the circulation, the draining renal lymph node, and distant (facial) lymph node at all points posttransplantation (Fig. 5). These results suggest that cotransplanted EPCs mediate the improvement of islet transplantation locally at the graft site in the early posttransplantation period.
Bone marrow-derived murine EPCs bind lectin and upregulate E-selectin. (A) Cultured bone marrow-derived EPCs were analyzed for lectin binding by flow cytometry. Unstained cells (dashed line) and fluorescein isothiocyanate-labeled lectin stained cells (filled histogram) are shown. (B) Upregulation of E-selectin was examined in EPCs cultured in the presence of tumor necrosis factor-α (TNF-α; 5 ng/ml) for 5 h with a representative of five experiments shown. Nontreated controls (dotted line) and TNF-treated cells (black histogram) were stained with anti-E-selectin. Isotype control on treated cells is shown (gray histogram). EPC cotransplantation improves cure rate of islet transplantation in a marginal mass model. Posttransplantation (day post-Tx) daily nonfasted blood glucose levels (BGL) of diabetic mice that received 200 islets (A) or 200 islets with 106 EPCs (B) under the kidney capsule. Each line represents one recipient mouse. #Animals humanely killed due to poor health or weight loss. Dotted line represents euglycemia (BGL < 11.1 mM). (C) Survival curve analysis shows improved rate of diabetes cure in animals receiving islets with EPCs (white squares; n = 12) compared to islets only (filled triangles; n = 10) (p = 0.002). (D) Average BGL (mean ± SEM) in groups that received islets only (filled triangles; n = 10) or islets with EPCs (white squares; n = 12). Euglycemic levels (BGL < 11.1 mM; shaded area) are shown. (E) Cured mice from each group were challenged at 28 days after transplant with 2 g/kg bodyweight of glucose injected intraperitoneally. Blood glucose levels were measured at various timepoints postinjection. Mean BGL ± SEM are shown for groups bearing islet-only grafts (dashed line/triangle; n = 2) or cotransplanted islets and EPCs (black line/square; n = 11). Cage-mate controls (dotted line/circle; n = 6) are shown for comparison. (F) Detection of CFDA-labeled EPCs (green) under the kidney capsule of mice cotransplanted with islets and EPCs 7 days earlier. The engrafted islet mass is shown stained for insulin (red). Scale bar: 100 μm. EPCs alone do not improve glycemic control in diabetic mice. Posttransplantation (day post-Tx) daily nonfasted blood glucose levels (BGL) of diabetic mice that received 106 EPCs alone (A; n = 5) or phosphate-buffered saline (PBS) (B; sham, n = 2) under the kidney capsule. Each line represents one recipient mouse. Dotted line represents euglycemia (BGL < 11.1 mM). Tracking of transplanted EPCs in vivo. EPCs were not detected in the circulation following cotransplantation under the kidney capsule. Pretransplantation labeled EPCs were gated by forward/side scatter (FSC/SSC) (A) and examined for green fluorescence [carboxyfluorescein diacetate succinimidyl ester (CFDA)] at the flow cytometer. (B) Labeled EPCs (filled histogram) showed a high level of fluorescence postlabeling compared to unlabeled cells (unfilled histogram). (C) Detection of 100 CFDA-labeled cells spiked into a full mouse blood preparation. (D) Mice cotransplanted with islets and labeled EPCs under the kidney capsule did not have detectable levels of labeled cells in their draining renal lymph nodes, ipsilateral facial lymph nodes, or blood at days 1, 7, or 28 posttransplantation.



EPC-Derived Factors Regulate Islet Function and β-Cell Connexin Expression In Vitro
We investigated whether soluble EPC-derived factors modify islets in vitro (Fig. 6). Islets were cultured in EPC-conditioned medium for 3 days, and their glucose-stimulated insulin release response was measured. During culture, freshly harvested medium was delivered to the islets daily to ensure that consumed factors were replenished. By examining the absolute amount of insulin released in each phase of glucose stimulation (relative to total protein; Fig. 6A), it was apparent that EPC-conditioned islets had an increased level of basal insulin release (p < 0.05), which partially explains the decreased stimulation index found in EPC-conditioned islets (p < 0.01) (Fig. 6B). There were no significant differences in the amount of insulin secreted during either the first or second phase response to high glucose. High potassium, which elicits the release of remaining stored insulin granules, resulted in similar levels of insulin being released, suggesting there was no difference in insulin protein translation or processing. The data suggest that EPC-conditioned islets have altered insulin release kinetics, rather than differences in insulin protein production or packaging. To investigate the mechanisms underlying the EPC-associated increase in basal insulin release, a separated Transwell coculture experiment was performed, where islets were grown in the presence of, but not in physical contact with, EPCs for 3 days (Fig. 6C). During culture, islet phenotype appeared altered, with many of the cocultured islets exhibiting a more adherent/stretched appearance compared to islets cultured alone (Fig. 6D). To examine underlying gene expression changes, islets were processed for real-time PCR analyzing a number of genes known to play a role in intraislet cell signaling (Fig. 6E). Ecad and Cx36 are two β-cell surface molecules suggested to be important for cell–cell tethering in coordinated islet insulin release (7,17). VEGF-A is secreted by β-cells and is likely to play a role in communication with islet vasculature (43). The integrin β1 chain (ITGB1) and its interaction with extracellular matrix components, such as type 1 collagens (Col1A1) and fibronectin (Fn1), are important for islet β-cell function (12,34). There was no significant difference in the transcript levels of insulin, Ecad, VEGF, collagen, integrin-1β, or fibronectin. Cx36 expression was significantly decreased in cocultured islets compared to islets cultured alone (p < 0.04; 0.4 ± 0.05-fold). Interestingly, pharmacological blockade of Cx36 with the inhibitor mefloquine in normal mouse islets caused a similar perturbation in glucose-stimulated insulin secretion (data not shown) as to that observed in islets cultured with EPC-conditioned medium (Fig. 6B). These data indicate that soluble factors produced by EPCs in culture are able to modulate the expression of the β-cell surface molecule Cx36, which may result in the dysregulation of glucose-stimulated insulin release in vitro.
Soluble EPC-derived factors modify islets in vitro. (A) EPC-conditioned medium altered insulin secretion kinetics in islets. Islets were cultured in control (white bars) or EPC-conditioned (filled bars) medium for 3 days. Groups of 10 islets (n = 6–7 per group) were assayed for their basal (3 mM glucose for 1 h), first phase (20 mM glucose for 10 min), second phase (20 mM glucose for 50 min), and high K+ (70 mM potassium) insulin secretion responses. Bars represent mean±SEM secreted into the supernatant corrected for total protein (*p=0.05). Stimulation indices (first phase divided by basal; B) were decreased in islets cultured in EPC-conditioned medium compared to control islets (bars represent mean±SEM; **p = 0.01). (C) Diagrammatic representation of islet/EPC separated coculture system. Twenty-five islets per well were cultured in cell-impermeant Transwells (0.4-μm pores) above EPC monolayers for 3 days prior to RNA extraction. (D) The morphology of islets cultured with EPCs appeared stretched compared to islets cultured alone. Scale bar: 50 μm. (E) Gene expression levels were analyzed by quantitative RT-PCR in islets cultured alone (white bars) or in the presence of EPCs (black bars). Graph shows average fold change (±SEM) of mean gene expression values [normalized to β-2-microglobulin (B2M)] relative to islets only. *p<0.04 compared to islets alone. INS, insulin; Ecad, E-cadherin; Cx36, connexin 36; VEGF, vascular endothelial growth factor; Col1A1, collagen type 1 α 1; ItgB1, integrin β1; Fn1, fibronectin 1.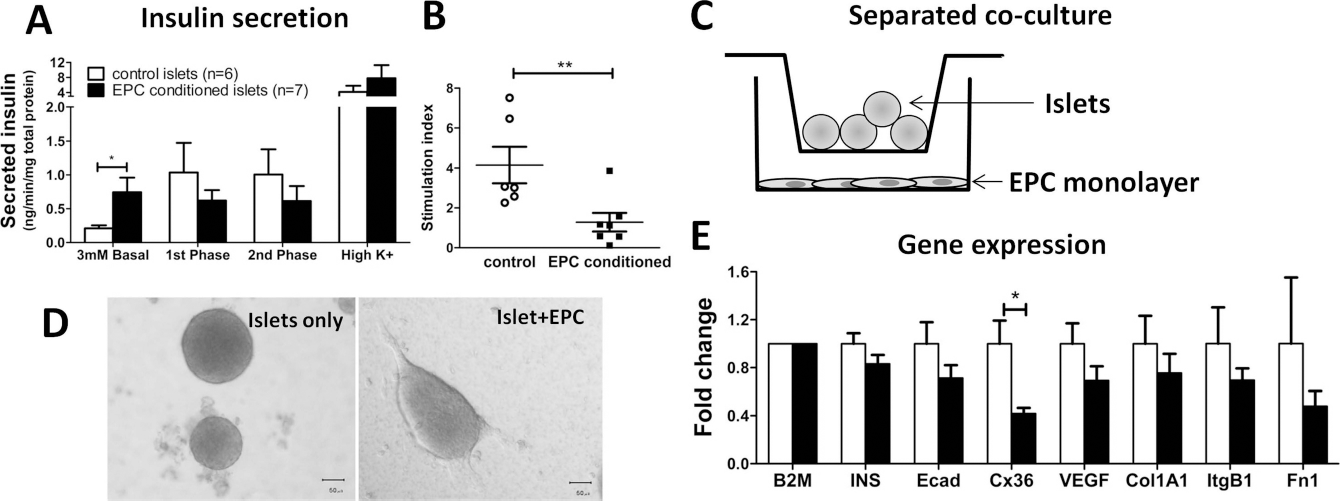
EPCs Modulate β-Cell Connexin 36 Expression via Soluble and Cell Contact Mechanisms
In order to examine whether Cx36 expression levels were also modulated by cell contact, EPCs and islets were cultured together for 4 h and processed for real-time PCR (Fig. 7). EPC doubling time ranges between 24 and 40 h (28), and the short coculture period was used to avoid the confounding influence of cell division and/or cell death. The coculture mixture was harvested and analyzed for gene expression. Gene levels are shown relative to the matched control: EPC-only and islet-only monocultures (matched for cell number) that were combined at the RNA extraction step. Genes examined included the β-cell-derived genes insulin, VEGF, and Cx36. While Cx36 expression is restricted to β-cells, intraislet endothelial cells express Cx43 and Cx45 (40). EPCs are known to express multiple angiogenic factors, including hepatocyte growth factor, which has particular relevance for β-cell survival (26). Thus, the expression of Cx43 and Cx45 and hepatocyte growth factor was measured in the cocultured samples. Again, Cx36 expression was significantly decreased during EPC contact coculture (p < 0.05; 0.3 ± 0.10-fold compared to controls) (Fig. 7B). Insulin expression was also reduced (p < 0.04; 0.6 ± 0.07-fold compared to postmix controls), suggesting that cellular contact between islet cells and EPCs may modify insulin expression at the mRNA level. Some grafted kidneys were analyzed for Cx36 mRNA expression at day 6–28 posttransplant, but there was no difference between kidneys bearing islets only compared to islets with EPC cotransplants (data not shown). Together, these data confirm that in vitro EPCs have the ability to modulate the expression of β-cell gap junction proteins including Cx36 and that this may occur rapidly when cells are in direct contact. Whether Cx36 is relevant for the in vivo function of islets in the context of transplantation will be an area of further study.
EPC contact modulates gene expression in islets in vitro. (A) Diagrammatic representation of islet/EPC contact coculture system. Islets (50 per well) were added to EPC monolayers and cultured for 4 h prior to RNA extraction. Control cultures comprised cell number matched monocultures (islet only or EPCs only) that were combined prior to RNA extraction step. (B) Gene expression levels of β-cell-derived or endothelial genes were analyzed by quantitative RT-PCR. Bars represent average fold change (±SEM) of mean gene expression values (normalized to B2M) in islet/EPC cocultures (black bars) relative to controls (white bars; monocultures combined at RNA extraction step). *p = 0.05 and **p = 0.04 compared to control cultures. HGF, hepatocyte growth factor.
Discussion
Intraislet endothelial cells have an increasingly appreciated role in supporting β-cell gene transcription, insulin secretion, survival, and proliferation. Here we have shown that intraislet endothelial cells are lost rapidly during islet culture and that exogenously delivered EPCs improve the cure rate of a marginal transplanted islet mass in a syngeneic diabetic mouse model. While potentially enhancing the engraftment and revascularization of transplanted islets in the early posttransplant period, these studies indicate that EPCs (or the soluble factors they produce) also directly impact β-cells, modulating the expression of the important β-cell molecule, Cx36.
Intraislet endothelial cells decreased rapidly during culture following islet isolation. These findings are unlikely to be due to dedifferentiation, or reduction in CD31 expression, because studies utilizing a GFP lineage-tagging technique have reported similar results (29). In the transplant setting, this decrease is likely to be important as donor islet-derived endothelial cells contribute to the formation of critical new blood vessels posttransplantation (5), along with the ingrowth of recipient-derived endothelial cells and bone marrow-derived cells (11,29). The consequent paucity of endothelial cells in cultured islets may in part explain their inferior graft survival compared to fresh islets (23,31). Surprisingly, a recent study examining islet engraftment into the anterior chamber of the eye found that fresh islets (containing relatively more endothelial cells) took longer to revert diabetes than 4-day cultured islets (30). However, compared to the kidney and the liver, the eye has a high oxygen tension, which may decrease the relative importance of revascularization responses for islet function at this site. Thus, the density of intraislet endothelial cells, and how quickly intraislet vascular connections become reestablished after transplantation, is likely to impact the function of pancreatic islets.
Possible mechanisms through which EPCs may support β-cell function and survival include via the production of soluble factors and through the direct interaction of cell surface molecules. We found that the culture of islets with EPC-conditioned media increased the amount of insulin released at basal glucose levels, but not at high glucose levels (Fig. 6). Thus, this change may represent a dysregulation of insulin release rather than an overall increase. These data conflict somewhat with those of Johansson et al. (21) who demonstrated that endothelial cell-derived soluble factors moderately enhance glucose-stimulated insulin release from rat islets in vitro. However, their study examined mature endothelial cells, and they postulated that endothelium-derived laminins were behind this improvement. It may be that mature endothelial cells produce more laminin than the immature EPCs tested here.
Other researchers have described an upregulation of islet-derived VEGF-A mRNA when cocultured with EPCs (22). There are two main differences between our experiments. Firstly, we utilized a syngeneic coculture system, deriving both EPCs and islets from mice, whereas Kang et al. examined the coculture of human umbilical cord blood-derived EPCs and murine islets. Second, our long-term culture experiments (3 days) were in physically separated cocultures, compared to using EPCs to coat the islets in culture. Thus, the robust upregulation of VEGF-A mRNA in response to EPCs described by Kang et al. (22) may be cell contact dependent.
We observed that extended culture with EPC-derived humoral factors, or short-term cell contact with EPCs, decreased the expression of Cx36 and increased basal insulin release in islets in vitro. Connexins are one group of molecules likely to be important for cell contact-mediated effects between β-cells and endothelial cells. Hexamers of these molecules form halves of gap junctions, with the ability to form complete channels with either connexins of the same (homojunctions) or different type (heterojunctions). Though Cx36 expression was initially thought to be restricted to neurons (10), it is the major connexin in pancreatic β-cells (37). Cx36 gap junctions allow the passage of ions and small molecules [in particular, Ca2+ and cyclic adenosine monophosphate (cAMP) (9)] between the cytoplasms of β-cells during synchronized glucose-induced Ca2+ oscillations (3,34). A particularly important function of Cx36 in islets is to extinguish Ca2+ elevations following membrane depolarizations (3,36,39). Thus, Cx36 connections within the islet allow less excitable β-cells to act as a buffer, suppressing electrical activity in their neighbors. Cx43 and Cx45 have been recently localized to endothelial cells within pancreatic islets (8,37,40), and it is tempting to entertain the possibility that β-cells and endothelial cells may communicate directly, via the exchange of electrical messages through heterotypic connexin junctions. Whether such connections are important in pancreatic islets remains to be established.
Herein we demonstrate that cotransplantation of EPCs with pancreatic islets improved the cure rate of a marginal mass of islets in a syngeneic murine transplant model. Despite having improved glucose control in the early transplant period, the ultimate function of transplanted islets in cured animals after 1 month was not significantly better than controls. Similarly, Nyqvist and colleagues (30) demonstrated moderately increased vascular density early posttransplantation when using fresh islets compared to cultured islets, but no difference at 1 month despite the long-term persistence of donor endothelial cells. Recent work from another group has shown that human EPC cotransplants speed the revascularization of porcine islets in diabetic nude mice (22). Importantly, the final engrafted β-cell mass was significantly better in cotransplanted mice, even though there was no difference in capillary density at 1 month posttransplantation. Thus, the main advantage of EPC cotransplantation may be to hasten the revascularization response posttransplantation. Whether this results in significantly improved graft function in the longer term may depend on the model used. Considering the large amount of cell death seen in clinically transplanted islets in the early posttransplant period, any enhancement of revascularization may translate to clinical improvement in transplantation outcomes.
In conclusion, the provision of EPCs during transplantation has the potential as a cell-based cotransplantation therapy to improve the engraftment of pancreatic islets. Within islets, it is likely that β/endothelial crosstalk is mediated via both humoral and cell contact- dependent mechanisms. While further mechanistic research is required, cell surface molecules involved in intraislet communication (such as Cx36) may represent important clinical targets in the future.
Footnotes
Acknowledgments
This research was supported by grants from Medvet Laboratories (2011/3001) and The Robinson Institute of the University of Adelaide (PRHJR10371). C.S. Bonder is a Heart Foundation Fellowship holder. C.F. Jessup is a holder of the Flinders University Vice Chancellor's Postdoctoral Research Fellowship. The authors thank Ms. Clare Mee for technical assistance and Dr. Michael Duffield for editorial assistance. The following author contributions are recognized: laboratory experiments for all in vitro and in vivo studies, collection of data, and manuscript writing (D.P.); laboratory experiments for in vivo studies and manuscript writing (D.R.-C.); laboratory experiments for in vivo studies (D.M.); laboratory experiments for in vitro studies (H.S.P.); laboratory experiments for in vitro studies (W.Y.S.); laboratory experiments for in vivo studies, data interpretation, and manuscript writing (C.J.D.); conception and design and manuscript writing (P.T.H.C.); conception and design, data interpretation, and manuscript writing (C.S.B.); financial support, conception and design, laboratory experiments for in vitro and in vivo studies, data collection and analysis, data interpretation, and manuscript writing (C.F.J.). The authors declare no conflict of interest.