
Abstract
Human multipotent adult progenitor cells (hMAPCs) are isolated from bone marrow with a more extensive expansion capacity compared to human mesenchymal stem cells (hMSCs) and with the ability to differentiate into endothelium. Like hMSCs, hMAPCs inhibit T-cell proliferation induced by alloantigens. In this study, we tested the interaction between hMAPCs and natural killer (NK) cells. We assessed the susceptibility of hMAPCs to NK cell-mediated lysis and the immunomodulation of hMAPCs on NK cell function during IL-2-driven stimulation and the cytolytic effector phase. Human MAPCs express the ligands PVR and ULBP-2/5/6, which are recognized by activating NK cell receptors. However, they also express MHC class I molecules, which induce inhibitory signals in NK cells. Freshly isolated NK cells at different effector:target ratios did not kill hMAPCs as assessed by an MTT and 51Cr-release assay, while hMAPCs impaired the cytotoxic activity of resting NK cells against the NK-sensitive K562 leukemia cell line. By contrast, IL-2-stimulated NK cells were capable of killing hMAPCs, and preactivated NK cells were not influenced during their cytotoxic effector function against K562 cells by hMAPCs. When added during the 6-day preactivation phase with IL-2, hMAPCs dose-dependently reduced NK cell proliferation in an IDO-dependent manner, but they did not influence the induction of cytotoxic capacity by IL-2. This study indicates that human MAPCs mutually interact with NK cells.
Keywords
Introduction
Stem cell-based therapy has become a promising tool to control immune responses. In the past decade, human mesenchymal stem cells (hMSCs) have been studied for their role as a cellular immunosuppressive cell population (25,26). MSCs are bone marrow-derived cells, which are capable of differentiating into chondrocytes, osteoblasts, and adipocytes (5,7). Clinical trials wherein hMSCs are infused intravenously are currently under way to evaluate their ability to prevent or suppress graft-versus-host disease (GvHD) in patients who underwent hematopoi-etic stem cell transplantation or to treat autoimmune diseases (11,12). The results of phase I and II clinical trials have demonstrated the feasibility and safety of in vivo use of these cells. The rationale for the use of hMSCs as an immunosuppressive cell population is based on a large number of studies that have shown that hMSCs inhibit T-cell responses in vitro (1,4,10,13), aside from affecting B-cells (2), dendritic cells (9,17), and natural killer (NK) cells in vitro (22-24).
NK cells are part of the innate immune system and play a key role in the immune defense against viral infections and in antitumor immune responses, based on their cytolytic function and production of proinflammatory cytokines. NK cell function is regulated by the balance between activating and inhibiting signals transduced by multiple cell surface receptors on NK cells (15). NK cell-mediated killing of a target cell requires the presence of activating ligands on the target cell interacting with activating receptors on the NK cells in combination with low or absent levels of major histocompatibility complex (MHC) class I molecules on the target cell, as the latter stimulate inhibitory receptors (14,16). MSCs are known to express high levels of MHC class I molecules (21). Both autologous and allogeneic MSCs can be lysed by activated NK cells. On the other hand, MSCs themselves can inhibit the cytotoxic activity and proliferation of NK cells (22-24).
Human multipotent adult progenitor cells (hMAPCs) are also bone marrow-derived stem cells (21). Compared with hMSCs, hMAPCs can be expanded more extensively than hMSCs. In addition, hMAPCs express lower levels of MHC class I molecules and differentiate robustly into endothelium both in vitro and in vivo in Matrigel plug assays. Like hMSCs, hMAPCs suppress allogeneic T-cell responses in vitro and block ongoing and secondary allogeneic T-cell responses and responses of memory T-cells (8). Their immunosuppressive potency, combined with their extensive proliferation potential, means that hMAPCs are a preferable alternative source for cell-based immunotherapy because a large cohort of patients can be treated with one single and well-defined batch of cells. Clinical trials with MultiStem®, the clinical-grade product of MAPCs, are being performed to test their ability to prevent acute GvHD, treat inflammatory bowel disease (IBD), and prevent rejection of liver grafts, aside from evaluating their ability to improve cardiac and neural function, when administered in the setting of acute myocardial infarction and stroke (www.ClinicalTrials.gov).
No systematic studies have been performed to address the interaction between hMAPCs and NK cells, so we here describe the NK cell function in the presence of hMAPCs and the hMAPC-mediated modulation of NK cell function during the interleukin (IL)-2-driven stimulation and cytolytic effector phase of NK cells.
Materials and Methods
Isolation and Culture of Stem Cells
hMAPC (n = 6) isolations were done by the Stem Cell Institute Leuven (SCIL, Leuven, Belgium) from bone fragments of four children (two male and two female between 5 and 15 years old; donors 1-4) undergoing orthopedic surgery or by ReGenesys (www.regenesys.eu; Heverlee, Belgium) from the bone marrow of two healthy volunteers [a 45-year-old male (donor 5) and a 30-year-old female (donor 6)], after obtaining informed consent in accordance with the guidelines of the Medical Ethics Committee of the University Hospitals Leuven. Isolation and culture of the cells were performed as previously described (20). Briefly, hMAPCs were generated by plating the total cell fraction at 0.5 × 106 cells/cm2 in medium consisting of 60% Dulbecco's modified Eagle's medium (DMEM) low-glucose (Gibco, Invitrogen, Carlsbad, CA, USA), 40% MCDB-201 (Sigma-Aldrich, St. Louis, MO, USA), supplemented with 50 nM dexamethasone, 10−4 M l-ascorbic acid, 1× insulin—transferrin—selenium (ITS), 0.5× linoleic acid—bovine serum albumin (LA-BSA) (all from Sigma-Aldrich), 1% penicillin/streptomycin (Gibco, Invitrogen), along with 2% Serum Supreme (Lonza BioWhittaker, Basel, Switzerland) and 10 ng/ml human platelet-derived growth factor (PDGF)-BB (R&D Systems, Minneapolis, MN, USA) and epidermal growth factor (EGF) (Sigma-Aldrich). MAPC cultures were maintained under hypoxic conditions (5% O2) at a density of 400 cells/cm2 and were split every 2 to 3 days. Clonal populations of hMAPCs isolated by SCIL were obtained through limiting dilution by plating five cells/well in a 96-well or 48-well plate (Corning, Corning, NY, USA) between passages 5 to 10. In some experiments, hMAPCs were treated with 100 U/ml interferon (IFN)-γ (Roche Diagnostics, Vilvoorde, Belgium) for 48 h.
Human MSCs were generated by ReGenesys from the bone marrow of the two adult hMAPC donors (donors 5 and 6) by plating the mononuclear fraction, obtained after Lymphoprep™ (Axis-Shield, Oslo, Norway) density gradient centrifugation, at 0.5 × 106 cells/cm2 in MSC growth medium containing DMEM high-glucose, 10% fetal calf serum (FCS), 100 IU/ml penicillin, and 100 μg/ml streptomycin (all from Lonza). MSC cultures were maintained at 5,000 cells/cm2, at normal oxygen level (20% O2), were split every 4 to 7 days, and were not clonally derived.
Isolation and Culture of Peripheral Blood Mononuclear Cells and NK Cells
All subjects donating blood for these experiments were healthy volunteers of both sexes, aged 20 to 50 years. Peripheral blood mononuclear cells (PBMCs) were separated by Ficoll-Hypaque (Axis-Shield) density gradient centrifugation (specific gravity, 1.077 g/ml). Untouched NK cells were negatively selected from PBMCs using the human NK cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany) according to the manufacturer's instructions. Purity ranged from 88% to 98%.
To obtain activated NK cells, PBMCs or purified NK cells were cultured for 6 days in Roswell Park Memorial Institute (RPMI) 1640 medium with 2 mmol/L l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all Lonza), and 10% autologous serum supplemented with 100 U/ml recombinant human IL-2 (TECIN; Hoffmann-La Roche, Nutley, NJ, USA) in T25 culture flasks (Greiner Bio-One, Wemmel, Belgium). PBMCs were cultured at 10 × 106 cells in 10 ml, while purified NK cells were cultured at 1 to 3 × 106 cells in 10 ml. To these cultures, nonirradiated hMAPCs were added in a ratio of 1:3 PBMCs or purified NK cells.
Flow Cytometry
hMAPCs, cultured PBMCs, or NK cells (1 × 105 for each) per sample were suspended in 100 μl phosphate-buffered saline (PBS; Lonza) supplemented with 10% heat- inactivated human serum (Lonza) to block nonspecific staining. Cells were subsequently surface stained with 5 μl fluorescence-conjugated specific monoclonal antibodies. The specifications of the antibodies used for flow cytometry are described in Table 1. Isotype control staining was performed. Acquisition was done using a FACSort or FACSCanto (BD Biosciences, Erembodegem, Belgium). For analysis of the samples, CellQuest Pro or BD FACSDiva software was used.
List of Monoclonal Antibodies Used for Flow Cytometry

CD, cluster of differentiation; HLA, human leukocyte antigen; PVR, poliovirus receptor; MICA/B, major histocompatibility complex (MHC) class I chain-related genes A and B; ULBP, UL16-binding protein; Ig, immunoglobulin; FITC, fluorescein isothiocyanate; PE, phycoerythrin.
Cytotoxicity Assays
The viability of hMAPC target cells was tested with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sigma, Bornem, Belgium) assay as previously described with some modifications (3). Briefly, target cells were seeded in a flat-bottomed 96-well cell culture plate (TPP, St. Louis, MO, USA) at a density of 5 × 104 cells/well and cultured overnight to become adherent to the plate. The next day, NK cells were added at different effector:target (E:T) ratios and cocultured for 24 h, after which the medium, together with the nonadherent cells, was removed. Subsequently, the target cells were rinsed with PBS at room temperature, and 100 μl of a 0.5 mg/ml MTT solution was added to the residual adherent cells. After 2 h of incubation, the MTT solution was removed, and 100 μl dimethyl sulfoxide (Merck, Darmstadt, Germany) per well was added to resolve the formazan produced in the viable cells. After stirring the plate for 5 min, optical density was measured at 570 and 620 nm using an ELISA reader (Thermo Labsystems, Franklin, MA, USA). The OD570-620 nm value was used as a measure for cell viability.
Cytotoxicity assays were also performed using a 4-h 51Cr-release method. Target cells were labeled with 100 μCi 51Cr/106 cells (51Cr, Perkin Elmer Life Sciences, Inc., Zaventem, Belgium) and seeded at 104 cells/well in round-bottomed 96-well plates (Greiner Bio-One). As control target cells, the human NK-sensitive and MHC class I-deficient K562 leukemia cell line was used (ATCC, Manassas, VA, USA). The lytic potential of NK cells was tested by coculturing cells at different E:T ratios. Saponin (Merck) was added to the target cells to measure the maximum release of 51Cr. Release of 51Cr was measured by a Topcount gamma counter (Packard Instrument Company, Meriden, CT, USA). The percentage cytotoxicity was calculated as [(experimental release - spontaneous release)/(maximal release - spontaneous release)] × 100.
To test the influence of hMAPCs during the lytic functioning of NK cells against K562 cells, nonirradiated hMAPCs were added at 1:2 ratio MAPC:NK at the beginning of the 4-h assay. We used the NK-resistant human cell line KM-H2 (Hodgkin disease-derived cell line; kindly provided by Dr. S. Fukuhara, Kyoto University, Kyoto, Japan) as control cells for hMAPCs in this assay.
Proliferation Assay
Responder purified NK cells (1 × 105) were stimulated with 100 U/ml exogenous recombinant IL-2 (TECIN; Hoffmann-La Roche). Irradiated (30 Gy) allogeneic hMAPCs were added at different suppressor:responder (S:R) ratios. NK cell proliferation was measured at day 6 by means of an 8-h pulse with 1 μCi/well [3H]thymidine (MP Biomedicals Europe, Illkirch, France). [3H]Thymidine incorporation was measured by using a liquid scintillation counter (Tri-Carb® 2100TR Liquid Scintillation Counter, PerkinElmer). The data were analyzed as mean counts per minute (cpm) of quadruplicate wells. The results are expressed as percent response related to the control response in the absence of hMAPCs.
To analyze the involvement of indoleamine 2,3-dioxygenase (IDO), prostaglandin E2 (PGE2), transforming growth factor (TGF)-β, and IL-10 as immunosuppressive mediators, we used their respective inhibitors: 200 μM/ml 1-methyl-tryptophan (1-MT; Sigma-Aldrich); 2 μg/ml indomethacin (Cayman Chemical Company, Ann Arbor, MI, USA), 50 μg/ml anti-TGF-β neutralizing mAb (R&D Systems, Abingdon, UK), and 2.5 μg/ml anti-IL-10 plus 2.5 μg/ml anti-IL-10 receptor mAb (both from R&D Systems).
Statistical Analysis
Statistics were calculated with Prism software 5.0 (GraphPad Software, Inc., San Diego, CA, USA). Statistical significance was calculated by paired or unpaired t tests for comparisons between two groups and by one-way ANOVA with Dunnett's post hoc test for comparisons between three or more groups. Values of p < 0.05 were considered significant.
Results
Human MAPCs Express Ligands of Activating NK Receptors
To assess whether hMAPCs might be susceptible for NK cell-mediated lysis, we first analyzed hMAPCs for the expression of ligands recognized by activating and inhibitory NK cell receptors. Table 2 shows the expression level of ligands of NK cell receptor D (NKG2D) [UL16-binding proteins (ULBPs) and MHC class I chain-related genes A and B (MICA/B)] and DNAX accessory molecule-1 [DNAM-1; cluster of differentiation 226 (CD226)] [Nectin-2 and poliovirus receptor (PVR; CD155)] triggering receptors and of inhibitory MHC class I molecules (HLA-ABC and nonclassic HLA-E) in five hMAPC populations (donors 1-5). Flow cytometric analysis showed that all hMAPC populations are dimly positive for HLA-ABC [mean fluorescence intensity (MFI) ± SEM for the five hMAPC populations: 1,992 ± 246] and negative for HLA-E. hMAPCs expressed PVR (8,540 ± 150) and low levels of ULBP-2/5/6 (1,173 ± 123), but generally did not express MICA/B, Nectin-2, ULBP-1, and ULBP-3. Upon stimulation with IFN-γ, expression of MHC class I molecules was upregulated, whereas the expression of the activating ligands remained unchanged.
NK Cell Receptor Ligand Expression by hMAPCs
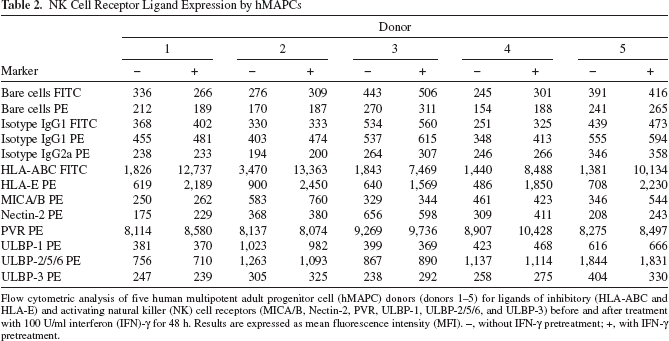
Flow cytometric analysis of five human multipotent adult progenitor cell (hMAPC) donors (donors 1-5) for ligands of inhibitory (HLA-ABC and HLA-E) and activating natural killer (NK) cell receptors (MICA/B, Nectin-2, PVR, ULBP-1, ULBP-2/5/6, and ULBP-3) before and after treatment with 100 U/ml interferon (IFN)-γ for 48 h. Results are expressed as mean fluorescence intensity (MFI). -, without IFN-γ pretreatment; +, with IFN-γ pretreatment.
Resting NK Cells Do Not Kill hMAPCs But Are Blocked in Their Cytolytic Function by hMAPCs
The combination of expression of ligands of activating NK cell receptors together with low levels of MHC class I molecules on hMAPCs suggests that hMAPC target cells might be killed by allogeneic NK cells. To investigate whether hMAPCs are susceptible to NK cell-mediated lysis, freshly isolated NK cells were cocultured with allogeneic hMAPCs at E:T ratios of 1:1 to 8:1 during 24 h. Coculture was followed by an assessment of hMAPC viability using the MTT assay. Human MAPCs were not killed by resting allogeneic NK cells (Fig. 1A). We confirmed these findings using a chromium release assay. This revealed that hMAPCs were only minimally lysed by resting allogeneic NK cells even at higher E:T ratios (Fig. 1B). Subsequently, to investigate whether hMAPCs could interfere with the effector function of resting NK cells, we added hMAPCs at the beginning of a 51Cr-release assay of freshly isolated NK cells against K562 target cells. As shown in Figure 2A, hMAPCs impaired the cytotoxic activity of resting NK cells (mean ± SEM% 51Cr-release: 44.81 ± 4.97% vs. 60.32 ± 3.40%). To exclude the possibility of “cold target inhibition” by hMAPCs, the NK-resistant cell line KM-H2 was used as a modulating cell line instead of hMAPCs. The resistance of KM-H2 cells to NK cell-mediated killing was verified (data not shown). Addition of KM-H2 cells at the beginning of a 51Cr-release assay of freshly isolated NK cells against K562 target cells did not significantly influence the cytotoxic activity of the NK cells, in contrast to hMAPCs (Fig. 2B).

Human MAPCs are not killed by resting NK cells. (A) Freshly isolated natural killer (NK) cells (n = 3) were cocultured with allogeneic human multipotent adult progenitor cell (hMAPC) target cells (n = 2; donors 5 and 6) at effector:target (E:T) ratios of 1:1 to 8:1 during 24 h. Coculture was followed by an assessment of hMAPC viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Data are expressed as percentage (mean ± SEM) of target cell viability (optical density) with respect to target cell viability without coincubation of NK cells (100%). Statistical significance was tested when compared with control condition without NK cells. One-way ANOVA with Dunnett's post hoc test was used, ns = not significant. (B) Freshly isolated NK cells were cocultured in a standard 51Cr-release assay with K562 cells or allogeneic hMAPCs at different E:T ratios of 50:1 to 25:1. Data are expressed as mean ± SEM percentage 51Cr-release of four different experiments with four different NK cell donors and three different hMAPC donors (donors 1, 5, 6).

Impaired cytotoxic activity of resting NK cells in the presence of hMAPCs. (A) Results of cytotoxic activity of freshly isolated NK cells against K562 target cells (E:T 25:1) in the absence (control) or presence (+hMAPCs) of allogeneic hMAPCs at suppressor:responder (S:R) ratio of 1:2. Results are expressed as mean ± SEM percentage 51Cr-release of 10 experiments, in which six different NK cell donors and two different hMAPC donors (donors 5 and 6) were used. Statistical significance was calculated with the paired t test. **p < 0.01. (B) Results of cytotoxic activity of freshly isolated NK cells against K562 target cells (E:T 25:1) in the absence (control) or presence of allogeneic hMAPCs (+hMAPCs) or NK-resistant KM-H2 cells (+KM-H2) at S:R ratio of 1:2. Results are expressed as mean ± SEM percentage 51Cr-release of four experiments, in which four different NK cell donors and two different hMAPC donors (donors 5 and 6) were used. Statistical significance was calculated with ANOVA and Dunnett's post hoc test. ns, not significant, *p < 0.05.
Activated NK Cells Lyse Allogeneic hMAPCs
The adoptive transfer of hMAPCs is aimed at controlling immune responses. The local inflammatory environment wherein hMAPCs reside upon injection will probably lead to activation of the regional NK cell population. To address the interaction between activated NK cells and allogeneic hMAPCs, we activated NK cells with rIL-2 for 6 days and tested their ability to kill allogeneic hMAPC target cells. The MTT viability assay revealed that preactivated NK cells were capable of killing allogeneic hMAPCs (Fig. 3A). We confirmed these findings using a chromium release assay, showing that the chromium release of hMAPCs as target cells for activated NK cells was similar to the control condition using K562 cells (Fig. 3B), indicating that hMAPCs can be killed by activated NK cells. However, pretreatment of hMAPCs with 100 U/ml IFN-γ for 48 h, leading to an upregulation of HLA-ABC and HLA-E expression (Table 2), rendered those IFN-γ-pretreated hMAPCs resistant to cytotoxic lysis by activated NK cells (6.59 ± 2.80% 51Cr release vs. 21.29 ± 5.55% 51Cr release) (Fig. 3C).
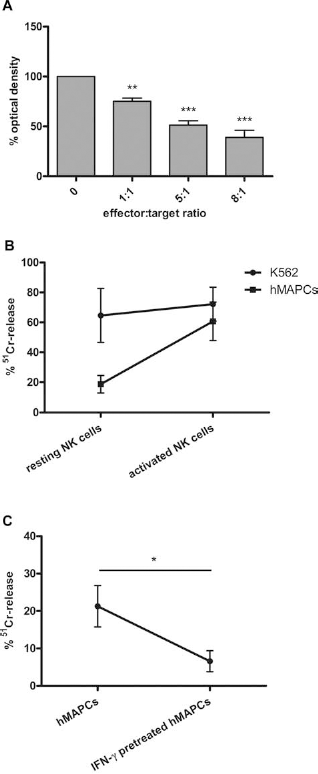
Human MAPCs are killed by activated NK cells. (A) NK cell populations (n = 2) that were exposed to 100 U/ml recombinant interleukin (rIL)-2 for 6 days were cocultured with allogeneic hMAPC (n = 2; donors 5 and 6) target cells at E:T ratios of 1:1 to 8:1. Coculture for 24 h was followed by an assessment of hMAPC viability using the MTT assay. Data are expressed as percentage (mean ± SEM) of target cell viability (optical density) with respect to target cell viability without coincubation of NK cells (100%). Statistical significance was tested when compared with control condition without NK cells. One-way ANOVA with Dunnett's post hoc test was used, ***p < 0.001, **p < 0.01. (B) Cytotoxic activity measured by 51Cr-release assay of freshly isolated NK cells (resting NK cells) and NK cell populations cultured for 6 days with rIL-2 (activated NK cells) against K562 or allogeneic hMAPC target cells at an E:T ratio of 25:1. Data are expressed as mean ± SEM percentage 51Cr-release of five different experiments with three different NK cell donors and three different hMAPC donors (donors 1, 5, 6). (C) Cytotoxic activity measured by 51Cr-release assay of activated NK cell populations cultured for 6 days with rIL-2, coincubated with allogeneic hMAPCs or interferon (IFN)-γ pretreated hMAPCs at an E:T ratio of 25:1. Data are expressed as mean ± SEM percentage 51Cr release of five different experiments with three different NK cell donors and three different hMAPC donors (donors 1, 5, 6). Statistical significance was calculated with the unpaired t test, *p < 0.05.
In addition, we tested the ability of hMAPCs to influence the cytotoxic function of activated NK cells during their effector phase. Therefore, a standard 51Cr-release assay was performed using NK cells that were cultured for 6 days in the presence of 100 U/ml rIL-2, and cytotoxic activity against K562 target cells was tested at an E:T ratio of 25:1 in the presence or absence of allogeneic hMAPCs (ratio hMAPC:NK cell 1:2). We noticed that the presence of hMAPCs reduced the percentage of 51Cr release (41.60 ± 3.12%) compared to the conditions cultured in the absence of hMAPCs (64.80 ± 4.17%) (Fig. 4). However, because activated NK cells have been shown to kill hMAPCs as well, we could not exclude that the reduction in 51Cr release was due to competition between hMAPCs and K562 cells as both cell populations are known target cells for the activated NK cells. To test this hypothesis, a 51Cr-release assay was performed using rIL-2 activated NK cells against K562 cells (E:T 25:1) in the presence or absence of extra unlabeled or 5ICr-labeled K562 cells or hMAPCs (both 1:2 ratio to NK cells). In comparison to the control condition, the percentage 51Cr release decreased with extra addition of unlabeled K562 cells or hMAPCs, while the percentage clearly increased with extra addition of labeled K562 cells or hMAPCs (data not shown). Addition of supernatant of cultured hMAPCs did not influence the cytotoxic function of rIL-2-activated NK cells. These observations imply a cold target inhibition effect.

Impaired cytotoxic activity of activated NK cells in the presence of hMAPCs. Results of cytotoxic activity of rIL-2-activated NK cells against K562 target cells (E:T 25:1) in the absence (control) or presence (+hMAPCs) of allogeneic hMAPCs at S:R of 1:2. Results are expressed as mean ± SEM percentage 51Cr release of four experiments, in which two different NK cell donors and two different hMAPC donors (donors 5 and 6) were used. Statistical significance was calculated with the paired t test. ***p < 0.001.
Human MAPCs Inhibit IL-2-Induced Proliferation of Allogeneic NK Cells in an IDO-Dependent Manner
We finally assessed whether hMAPCs have a similar suppressive effect on NK cell proliferation as they have on T-cell proliferation. NK cells were stimulated with rIL-2 (100 U/ml) in the presence or absence of irradiated allogeneic hMAPCs at different S:R ratios and evaluated with [3H]thymidine incorporation after 6 days. As shown in Figure 5A, human MAPCs dose-dependently suppressed IL-2-induced proliferation of highly purified NK cells. The expansion of NK cells (defined as % CD3-CD56+ cells) was also blocked when total PBMC fractions were cultured in medium supplemented with exogenous rIL-2 for 6 days in the presence of hMAPCs (10.64 ± 2.77% CD3-CD56+ cells) compared to similar cultures of PBMCs in the absence of hMAPCs (37.57 ± 3.59% CD3-CD56+ cells) (Fig. 5B). Of note, in this set of experiments, hMSCs from the same donor as the hMAPCs were available. The decrease in the expansion of NK cells was less pronounced when PBMCs were activated with rIL-2 in the presence of hMSCs (22.70 ± 8.76% CD3-CD56+ cells), compared to the condition with hMAPCs.

hMAPCs dose dependently suppress IL-2-induced NK cell proliferation. (A) Freshly isolated NK cells were cultured during 6 days in medium supplemented with 100 U/ml rIL-2 in the absence or presence of allogeneic irradiated (30 Gy) hMAPCs at different S:R ratios. The proliferative response was measured on day 6 by [3H]thymidine incorporation. Results are expressed as mean ± SEM percentage proliferation relative to control cultures in the absence of hMAPCs of six experiments in which five different NK cell donors and three different hMAPC donors (donors 1, 5, 6) were used. Statistical significance was calculated with one-way ANOVA with Dunnett's post hoc test, **p < 0.01, *p < 0.05. (B) Total peripheral blood cell (PBMC) fractions were cultured in medium supplemented with exogenous rIL-2 for 6 days in the absence (upper left) or presence of human mesenchymal stem cells (hMSCs; upper right) or hMAPCs (lower left) from the same donor at S:R ratio of 1:3. The expansion of NK cells [percentage cluster of differentiation 3 negative cluster of differentiation 56 positive (CD3-CD56+) cells] in the total PBMC fraction was afterwards analyzed by flow cytometry. One out of three representative experiments is shown in the flow cytometry plots. In the bar graph (lower right), results from three different experiments with three different PBMC donors and two hMSC/hMAPC donors (donors 5 and 6) were pooled and expressed as mean ± SEM percentage CD3-CD56+ cells. Statistical significance was calculated with one-way ANOVA with Dunnett's post hoc test, ns = not significant, *p < 0.05.
The immune modulatory mechanism of hMAPCs is mediated at least in part via a soluble factor (8), so we performed some functional tests to identify the responsible mediator. Blocking IDO with 1-MT completely neutralized the inhibitory activity of hMAPCs on IL-2-induced NK cell proliferation, suggesting IDO as a mediator of the immune modulation by hMAPCs (Fig. 6). Addition of monoclonal antibodies neutralizing IL-10 plus IL-10R, or monoclonal antibodies to neutralize TGF-β, or addition of indomethacin to neutralize PGE2 synthesis did not change the immunomodulatory properties of hMAPCs on IL-2-induced NK cell proliferation. None of these molecules mediated the immune modulatory role of hMAPCs on the cytotoxic activity of resting NK cells against K562 cells (data not shown).

The suppressive effect of hMAPCs on NK cell proliferation is dependent on IDO activity. Freshly isolated NK cells were cultured during 6 days in medium supplemented with 100 U/ml rIL-2 in the absence or presence of irradiated hMAPCs at S:R ratio of 1:2 without (control) or with addition of 2 μg/ml indomethacin, 200 μM/ml 1-methyl tryptophan (1-MT), 2.5 μg/ml anti-IL-10 plus 2.5 μg/ml anti-IL-10 receptor (R) mAb, or 50 μg/ml anti-transforming growth factor (TGF)-β neutralizing mAb. The proliferative response was measured on day 6 by [3H]thymidine incorporation. Data are expressed as mean ± SEM percentage proliferation relative to control cultures in the absence of hMAPCs of three experiments in which two NK cell donors and two hMAPC donors (donors 5 and 6) were used. The average counts per minute (cpm) of the cultures without hMAPCs for all conditions were all similar to the control condition without hMAPCs (average cpm: 13,855). IDO, indoleamine 2,3-dioxygenase.
In the next set of experiments, total PBMC fractions were cultured for 6 days with rIL-2 in the presence or absence of nonirradiated hMAPCs, and the cytolytic function of the NK cells was subsequently measured against K562 cells. As shown in Figure 7A, the lytic activity was strongly diminished when the whole PBMC population was stimulated with rIL-2 in the presence of hMAPCs compared to the control condition without hMAPCs. We did not see any influence on the cytotoxic activity of the NK cells when PBMCs were activated with rIL-2 in the presence of hMSCs from the same hMAPC donors. The cytolytic activity in these experiments was influenced by the inhibition of NK cell proliferation, thereby influencing the net E:T ratio during the subsequent effector phase, so similar experiments were performed with purified NK cells as responder and effector cells. After stimulation of purified NK cells with rIL-2 in the presence of nonirradiated hMAPCs, the cells were adjusted prior to the cytotoxicity assay. In this condition, we could demonstrate that the cytolytic function of NK cells was retained after rIL-2 activation in the presence of hMAPCs, in spite of the blocked proliferative response (Fig. 7B). Of note, addition of irradiated hMAPCs in this experimental condition yielded similar results (data not shown).

Suppression of NK cells by hMAPCs during activation with rIL-2. (A) After 6-day culture with rIL-2 in the presence or absence of hMAPCs or hMSCs (S:R ratio of 1:3) during the activation phase, the total PBMC fraction was subsequently cocultured with K562 leukemia cell line at E:T ratios of 50:1 and 25:1 during the effector phase. Specific K562 lysis was measured with 51Cr release. Data are expressed as mean percentage 51Cr release of three independent experiments using three different PBMC donors and two different hMAPC/hMSC donors (donors 5 and 6). (B) After culture of 6 days with rIL-2 in the presence or absence of hMAPCs (S:R ratio of 1:3) during the activation phase, purified NK cells were cocultured with K562 leukemia cell line at E:T ratios of 50:1 and 25:1 during the effector phase of NK cells. Specific K562 lysis was measured with 51Cr release. Data are expressed as mean ± SEM percentage 51Cr release of eight experiments using five different NK cell donors and three different hMAPC donors (donors 1, 5, 6).
Discussion
In the present study, we report on the interaction between hMAPCs and NK cells. Resting NK cells do not kill hMAPCs and are blocked in their cytolytic function by hMAPCs. hMAPCs block IL-2-induced NK cell proliferation but not their subsequent effector function. On the other hand, activated NK cells kill hMAPCs, unless the latter were preincubated with IFN-γ and are no longer blocked by hMAPCs during their cytotoxic functioning. Hence, there is a balance in the mutual interaction between hMAPCs and NK cells depending on the activation state of the NK cells and the priming of hMAPCs.
Our data on the NK cell susceptibility of allogeneic hMAPCs are in accordance with published reports on hMSCs (18,22,24). hMAPCs express lower levels of MHC class I molecules compared to hMSCs (8,21), and also express ligands for activating NK cell receptors on its surface. To trigger a NK cell, failure to recognize appropriate inhibitory ligands on a target cell is mandatory. MSCs express high levels of MHC class I molecules, so it was hypothesized that MSCs would escape NK cell-mediated lysis. Indeed, published data showed that MSCs resist lysis of alloreactive resting NK cells. However, activated NK cells are capable of killing allogeneic and autologous MSCs (6,22,24). The data suggest that the interaction between MHC class I-specific inhibitory receptors on NK cells and the MHC class I molecules on the MSCs is not sufficient to protect MSCs from lysis. The susceptibility of hMAPCs for activated NK cells may hamper their survival in vivo. hMAPCs will be used as an off-the-shelf stem cell product and will be consequently of third-party origin; it can therefore be hypothesized that in an inflammatory environment, which is the situation in the case of GvHD or ischemia, nearly all hMAPCs will be killed. This however might, at least in part, be counteracted by the inflammation-induced upregulation of MHC class I molecules on hMAPCs. Indeed, for hMSCs, Spaggiari et al. reported that the upregulation of MHC class I molecules on hMSCs due to stimulation with IFN-γ rendered these cells resistant to NK cell-mediated lysis (24). We were able to document similar results for hMAPCs. Based on these findings, we can hypothesize that the final outcome of the interaction between hMAPCs and NK cells in vivo will depend on the local inflammatory environment.
The presence of hMAPCs impaired the cytolytic potential of resting NK cells in vitro. hMAPCs were not killed by resting NK cells, so the reduced cytotoxic effect of resting NK cells in the presence of hMAPCs was not due to competition of target cells. We confirmed this hypothesis by using NK-resistant KM-H2 cells instead of hMAPCs as modulating cells in some experiments. The question whether hMAPCs were able to block the cytotoxic function of rIL-2-activated NK cells could not be answered in our experiments, as activated NK cells were killing both the K562 target cells and hMAPCs.
hMAPCs suppressed rIL-2-induced proliferation of NK cells during stimulation with rIL-2 for 6 days but not their cytotoxic function. The mechanism responsible for the immunosuppression of hMAPCs is not yet completely understood. Several candidate molecules have been proposed as the soluble immunosuppressive factor produced by hMSCs, although data are contradictory because of different experimental designs to study immunosuppression (1,4,13,19). In our hands, blockage of TGF-β, IL-10, or PGE2 synthesis did not influence the suppressive effect of human MAPCs on rIL-2-induced NK cell proliferation. Addition of 1-MT to cocultures of freshly isolated NK cells with hMAPCs resulted in a loss of the hMAPC-mediated suppressive effect on IL-2-mediated NK cell proliferation, confirming the role of IDO as one of the responsible mediators. None of these mediators was responsible for the modulation during the cytotoxic effector phase of resting NK cells. Our data are in accordance with previously published results on T-cell alloreactivity (8).
The present study was primarily aimed to study the mutual interaction between hMAPCs and NK cells. Related to sensitivity to NK cell-mediated killing, modulation of NK cell proliferation, and mechanism of immunomodulation, hMAPCs did not differ in function from hMSCs as described in literature. Nevertheless, some differences in immunomodulatory action between hMAPCs and hMSCs were found compared to literature and from own experiments. First of all, the modulatory effects of hMAPCs during the rIL-2-induced stimulation phase of NK cells in our study were different compared to hMSCs in the study by Spaggiari et al. (23). In the latter study, coculture of purified NK cells with irradiated hMSCs for 6 days in the presence of rIL-2 did not only strongly reduce the rIL-2-induced proliferation but also inhibited the cytotoxic activity of the purified NK cells. Next, in our hands, hMSCs became available from the same donor as hMAPCs only for the experiments on rIL-2-induced stimulation of PBMC populations. Enrichment of NK cells upon rIL-2 stimulation in the PBMC cultures was blocked more by hMAPCs than by hMSCs. As a consequence, the subsequent NK potency of these stimulated PBMCs was also reduced in the cultures in the presence of hMAPCs but less in the presence of hMSCs. Third, Rasmusson et al. demonstrated that hMSCs did not influence the specific K562 lysis of resting NK cells (18), whereas hMAPCs in our study reduced the cytotoxic function of resting NK cells when cocultured during the effector phase.
The fate of hMAPCs after injection into patients is not yet understood. In this study, we demonstrated that hMAPCs interact with NK cells in vitro. The final outcome of this interaction in vivo will depend on the inflammatory microenvironment both affecting the activation state of the NK cells and also the MHC expression on hMAPCs. Our data add to the understanding of clinical results of currently running trials using adoptive transfer of hMAPCs. Further in vivo studies with patients treated with hMAPCs should be done to better understand the fate and function of hMAPCs as immune-modulating stem cell population.
Footnotes
Acknowledgments
The authors would like to thank Lieve Coorevits, Emily Dekimpe, Elke Nackers, and Anaïs Van Hoylandt, KU Leuven, for their excellent technical assistance. We would also wish to thank Vik Van Duppen, KU Leuven, for his help with the flow cytometry and Omer Rutgeerts, KU Leuven, for his help with the 51Cr-release assay. This work was supported by the Center of Excellence (EF/05/011) funding KU Leuven, an Odysseus award, research funding from Athersys Inc., and a grant from the European Commission (EC-FP6-STREP-STROKEMAP) to Catherine M. Verfaillie. Valerie D. Roobrouck was funded by a grant from the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT Vlaanderen). Stefaan W. Van Gool is senior clinical investigator of FWO Vlaanderen. Catherine M. Verfaillie is a consultant to Atherys Inc., and research funds were obtained from Athersys Inc. Jef Pinxteren is manager and head R&D of ReGenesys. Sandra A. Jacobs designed and performed experiments, collected and analyzed results, wrote and revised the manuscript. Jeroen Plessers designed and performed experiments, collected and analyzed results, wrote and revised the manuscript. Jef Pinxteren designed and performed experiments, revised the manuscript. Valerie D. Roobrouck designed and performed experiments, revised the manuscript. Catherine M. Verfaillie revised the manuscript. Stefaan W. Van Gool did conception and design, analyzed and interpreted data, wrote and revised the manuscript. Authors declare no additional potential conflicts of interest.