
Abstract
A positive effect on the future development of cells, which have been cooled to low suprazero temperatures and then thawed, has been observed before and is not new. The aim of this study was to test the effectiveness of postthawing culture of human ovarian tissue, which was either frozen just after operative removal or cooled after removal to 5°C for 24 h before cryopreservation. Ovarian fragments from six patients were divided into small pieces in the form of cortex with medulla and randomly divided into the following four groups: Group 1 consisted of pieces that just after removal had been cultured in vitro for 8 days in a big volume of medium with mechanical agitation; Group 2 included pieces cooled after operation to 5°C for 24 h and then cultured in vitro for 8 days; Group 3 was comprised of pieces frozen–thawed just after operation and then cultured for 5 days in the chorioallantoic membrane (CAM) culture system; and the pieces in Group 4 were cooled after operation to 5°C for 24 h, frozen–thawed, and then cultured in the CAM system for 5 days. The effectiveness of the tissue culture was evaluated by the development of follicles and by the intensiveness of proliferation in the tissue (by expression of cytokeratin and Ki-67). For Groups 1, 2, 3, and 4, the mean densities of follicles per 1 mm3 was 12.9, 12.2, 12.4, and 16.1, respectively (p1–2 > 0.1; p3–4 < 0.05). For these groups, 87%, 95%, 71%, and 84% of the preantral follicles were morphologically normal (p1–2, 3–4 < 0.05). The immunohistochemical analysis showed increased proliferation after cooling of fresh and cryopreserved tissue. Long-term (24 h) cooling of ovarian tissue to 5°C before cryopreservation increases the viability of the cells in the tissue after thawing. Additionally, the efficacy of the CAM system for the culture of thawed human ovarian tissue was demonstrated.
Introduction
Owing to the increasing effectiveness of cancer treatments and positive long-term prognosis for young women, the problem of postcancer infertility is playing an increasingly significant role because chemotherapy can be gonadotoxic and lead to the functional death of ovaries. Cryopreservation of ovarian tissue before cancer therapy with reimplantation after convalescence is the potential key solution to this problem (6,33,34).
Several cases reporting restoration of ovarian function after reimplantation of ovarian cortex in patients with premature ovarian failure after cancer treatment have been published since 1998. Now, pregnancies and babies born after retransplantation of frozen ovarian tissue have been reported (31).
Part of the ovarian tissue obtained before oncological treatment is used for routine histological observation, a mandatory procedure for monitoring and minimizing the risks associated with any future implantation of tissues that could be affected by metastases. After cryopreservation and storage, some ovarian tissue can be thawed and cultured in vitro in order to screen for the presence of metastases and to monitor follicles. The quality of follicles in the cultured tissue will indicate whether it is possible to restore a woman's reproductive function. At present, there are three ways to effectively determine the quality of the cryopreservation procedure using ovarian tissue from a given patient before the reimplantation treatment: (i) evaluation of follicles after postthawing xenotransplantation to severe combined immunodeficient (SCID) mice (9,19,24,25,27–29), (ii) in vitro culture in a large volume of culture medium and under constant agitation (10–12), and (iii) culture on embryonic chorioallantoic membrane (CAM) within a hen's egg (13,21).
The CAM system is an intermediate stage between in vitro culture and animal experiments, and it could be considered as an interface between in vitro and in vivo models (including xenotransplantation). Importantly, it does not raise ethical or legal questions, nor does it violate animal protection laws.
At present, the suprazero temperature (usually from 0°C to 7°C) storage method is a widely used technology in organ preservation. Low temperature retards cellular metabolism and thus reduces cellular oxygen demand and consumption. This furnished an approach to reduce tissue autolysis without the need for vascular perfusion.
The absence of a negative effect of hypothermic storage on domestic cat (35) and rat (36) ovaries has been noted. The positive effect of cooling on rat hypothalamus was reported (7).
Comparative data about the effect of long-term cooling of human ovarian tissues before cryopreservation on their viability after thawing are limited.
We chose two culture methodologies for the current investigation: the in vitro and the CAM system because the chorioallantoic membrane system is a borderline model, an intermediate stage between in vitro culture and animal experiments.
The aim of this study was to compare the effectiveness of postthawing culture of human ovarian tissue, which was either frozen just after removal (surgical operation) or cooled to 5°C for 24 h just after the operation and later, after this 24-h cooling, was frozen.
Materials and Methods
Full details of the study described in this article were approved by the Ethics Boards of Universities Cologne (permission 276-03 from 07.07.2003) and Ulm (permission 102/10 from 19.07.2010).
Written informed consents were obtained from all participants aged 18 and over involved in our study. On behalf of the patients under the age of 18, written consents were obtained from the next of kin.
Except where otherwise stated, all chemicals were obtained from Sigma (Sigma Chemical Co., St. Louis, MO, USA).
Tissue Collection, Dissection, and Distribution Into Groups
Informed consent was obtained from six female patients aged between 13 and 32 (22.1±5.0) years. According to approved protocol, 10% of ovarian tissue was used for patient-oriented research.
The basal medium used for manipulation of tissues (transport and dissection) was Leibovitz L-15 with 5% dextran serum substitute (Irvine Scientific, Santa Ana, CA, USA), referred to below as “basal medium.”
Fresh ovarian tissue fragments were transported from the surgical room to the laboratory within 20 min with temperature maintained at 32–34°C. Using tweezers (Bochem Instrumente GmbH, Weilburg, Germany) and scalpel No. 22 (B Braun Melsungen AG, Melsungen, Germany), ovarian fragments were dissected and divided into small pieces (1.5–2.0 × 1.0–1.2 × 1.0–1.2 mm) and cryopreserved as described below. The pieces were randomly divided into four groups:
Group 1: Pieces were cultured in vitro for 8 days after surgical operation (removal of these pieces).
Group 2: Pieces were cooled after operation to 5°C for 24 h and then cultured in vitro for 8 days.
Group 3: Pieces were frozen just after operation without cooling and then thawed and cultured for 5 days in a chorioallantoic membrane (CAM) culture system.
Group 4: Pieces were cooled after operation to 5°C for 24 h, then frozen, thawed, and cultured in the CAM system for 5 days.
Eight pieces in one experimental group were used for respective treatment (freezing and culture or only culture with or without cooling) to determine the quality of follicles and the degree of proliferation. After thawing, one half of the experimental pieces (n = 16) were dissected on 32 fragments (~1.0 × 1.0 × 1.0 mm) and in vitro cultured. The second half of the pieces were dissected on 48 fragments (0.5–1.0 × 0.5–1.0 × 1.0–1.2 mm) and CAM cultured. The volume of pieces for CAM culture is smaller than pieces for in vitro culture. After respective treatment and culture, 79 particles of ovarian cortex with medulla (32 after in vitro culture and 47 after CAM culture) were analyzed.
Cryopreservation (Freezing and Thawing)
Pieces of ovarian tissue were placed at room temperature in 20 ml of freezing medium composed of basal medium supplemented with 6% dimethyl sulfoxide, 6% ethylene glycol, and 0.15 M sucrose. Then pieces were put into a standard 1-ml cryostraw (MTG GmbH, Bruckberg, Germany) previously filled by freezing medium and frozen in a CTE 2104 freezer (Cryo-Technik-Erlangen, Hoechstadt, Germany). The freezing program consisted of the following five stages: (1) start temperature 22°C, (2) cooling from 22°C to 4°C at a rate of −5°C/min, (3) cooling from 4°C to −7°C at a rate of −1°C/min, (4) cooling from −7°C to −34°C at a rate of −0.3°C/min, (5) plunging into liquid nitrogen. It was noted that at −10°C there was a formation of crystals in the place of localization of ovarian pieces (this construction of freezer implies the autoseeding).
The procedure of thawing was achieved by holding the straws for 10 s at room temperature followed by immersion in a 100°C (boiling) water bath for 20 s and expelling the contents of the straw into the solution for the removal of cryoprotectants. The exposure time in the boiling water was visually controlled by the presence of ice in the medium; as soon as the ice was 2 to 1 mm apex, the straw was removed from the boiling water, at which point, the final temperature of the medium was between 4°C and 10°C. Within 5–10 s after thawing, the pieces from the straw were expelled into 10 ml of thawing solution (basal medium containing 0.5 M sucrose) in a 100-ml specimen container (Sarstedt, Nuembrecht, Germany). The stepwise dilution of cryoprotectants was achieved using the same principle as that used for saturation by ethylene glycol by Isachenko et al. (12). The container was placed on a shaker and continuously agitated at 200 osc/min for 15 min at room temperature. Stepwise rehydration of the tissue pieces for 30 min at room temperature was also performed using the same “dropping” methodology: slow addition of basal medium (see above) to the solution of sucrose with ovarian pieces. For “dropping,” we used 50 ml of basal medium in a 50-ml tube (Greiner Bio-One GmbH, Frickenhausen, Germany). The final sucrose concentration was 0.083 M, resulting in almost isotonic conditions. Finally, the pieces were washed thrice each in basal medium for 10 min and transferred for CAM culture.
In Vitro Culture
Individual thawed pieces of two experimental groups (Group 1: pieces immediately after operation and Group 2: pieces cooled after operation to 5°C for 24 h) were placed in 200-ml dishes for suspension culture (Cellstar, Greiner Bio-One GmbH) with 30 ml of AIM-V medium (Gibco/Invitrogen, Gaithersburg, MD, USA). They were incubated for 8 days at 37°C in 5% CO2 with 75 osc/min agitation using a rotation shaker.
CAM Culture
Fertilized eggs (n = 48) of White Leghorn chickens, purchased at a local hatchery and incubated at 37°C with 60% relative humidity, were prepared for implantation on day 4 of incubation. Standard microbiology assessment was performed to exclude subclinical infections. Preparation of the CAMs was performed essentially as previously described (3,11,13,16,17,21). Each egg was washed with warm 70% ethanol, after which a hole was drilled through the pointed pole of the shell (Fig. 1). The following day, part of the CAM of the embryo was exposed by peeling a 1.5- to 2.0-cm window in the shell. This window was covered with tape and the incubation continued. The CAM has two epithelial layers and, in its intact form, represents a “dry” impermeable barrier for all invasions, including ovarian fragments. For “connection” of ovarian pieces with the egg system, the latter must be open. For this, the upper peridermal part of the double epithelial layer was removed in each egg, leaving the basal layer intact. On day 10 of incubation, a silicone ring 0.5 mm thick with a 5-mm inner diameter (Sudhof Technik GmbH, Ulm, Germany) was placed on the membrane (Fig. 1). An ovarian piece was placed into this silicone ring using microsurgical forceps. Thereafter, the shell window was covered again, and the egg was replaced in the incubator. After 5 days of CAM culture, the viability of ovarian pieces was evaluated.
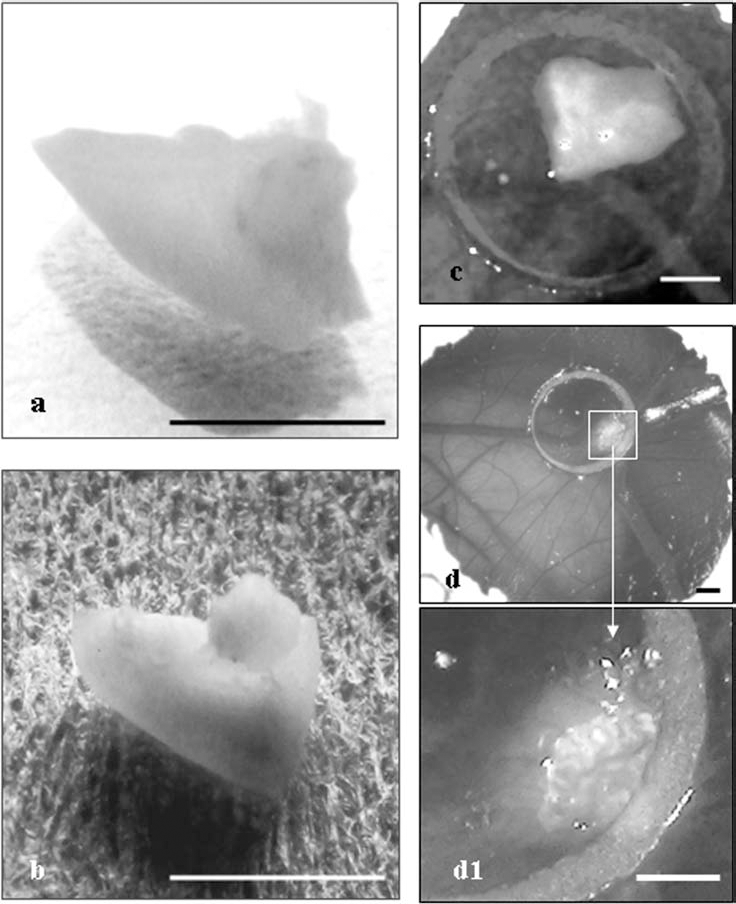
Ovarian pieces precooled to 5°C for 24 h. (a, b) Eight-day in vitro culture. (a) Piece after precooling to 5°C and before in vitro culture. (b) The same piece after 8 days in vitro culture. (c, d, d1) Five-day chorioallantoic membrane (CAM) culture. (c) Piece after 24 h precooling to 5°C, freezing and thawing, before CAM culture. (d, d1) The same piece after 5-day CAM culture. Scale bar: 1 mm.
Histology of Follicles
For histological investigation, the cultured tissue pieces were fixed in Bouin's solution, embedded in paraffin wax, serially sectioned at 4 μm, stained with hematoxylin/eosin, and analyzed under a microscope (400×, Olympus Co, Tokyo, Japan).
The number of viable and damaged follicles was counted. Before the counting of follicles, sections were coded and scored “blind.” To avoid overcounting of the same follicles, only the section with a visible oocyte nucleus was taken into account. Morphology of the follicles was evaluated on the basis of parameters previously described (26). Two types of preantral follicles were evaluated: (1) primordial follicles composed of an oocyte surrounded by a layer of flattened follicular cells and (2) primary and secondary follicles, which are similar to primordial follicles, but in which the oocyte is surrounded by one to two layers of spheroid granulosa cells.
The quality of follicles was graded on the scale from 1 to 3. A follicle of grade 1 is spherical in shape and contains a spherical oocyte, which is surrounded by an even distribution of granulosa cells and has a homogenous cytoplasm and slightly granulated nucleus, with condensed chromatin in the form of a dense spherical structure detectable in the center of the nucleus. A follicle of grade 2 has similar characteristics, but the oocyte is without condensed chromatin within the nucleus and is often irregular in shape; the surrounding granulosa cells can be flat and pulled away from the edge of the follicle. A follicle of grade 3 contains a misshapen oocyte with or without nuclear vacuolation; theca and granulosa cells are separated from the edge of the follicle, and the partly or fully disrupted granulosa have pyknotic nuclei. Follicles of grades 1 and 2 were denoted as normal, and those of grade 3 were denoted as degenerated. Examples of the different follicular degenerations can be observed elsewhere [e.g., see Isachenko et al. (11,13)].
Immunohistology for Proliferation
For these investigations, ovarian pieces were fixed in 4% paraformaldehyde, embedded in paraffin wax, and serially sectioned at 4 μm. After deparaffinization, slides were processed for staining for human cytokeratin and Ki-67 (antibodies and autostainer from Dako, Hamburg, Germany) according to manufacturer's protocol. FLEX monoclonal mouse anti-human Ki-67 antigen, clone MIB-1 and EnVision, FLEX Target Retrieval Solution, low pH, Code K8005, as well as FLEX monoclonal mouse anti-human cytokeratin 5/6, clone D5/16 B4, Code IS780 and EnVision, FLEX, high pH, Autostainer Plus, Code K8010 were used for immunization and visualization. Images were digitally scanned at 50× magnification with an Axiophot microscope (Carl Zeiss, Jena, Germany) and recorded.
Our scoring system was similar to that described by Abir et al. (1,2). The intensity of immunoreactivity with different monoclonal antibodies was subjectively assessed as follows: lack of immunoreactivity, immunoreactivity low, medium, and high.
Statistical Analysis
Integrity rate of tissue after treatment was evaluated by ANOVA. Various characteristics were summarized by mean and SD within groups. The level of statistical significance was set at p < 0.05.
Results
The survival rate of chick embryos was 97.9% (47 of 48).
After 8 days of in vitro culture, as well as 5 days after transfer onto the extraembryonic vascular system of the chorioallantoic membrane, the ovarian fragments were observed to have developed a spherical shape (Fig. 1).
After completion of CAM culture (on the fifth day), the propagation of ovarian pieces in the mesenchymal layer of the membrane was noted (Fig. 1).
Histology of Follicles
Histological examination after 8 days of in vitro and 5 days of CAM culture of ovarian pieces of Group 2 (24-h postoperation cooling to 5°C and following 8 days in vitro culture), Group 3 (cryopreservation just after operation without cooling and following 5 days CAM culture) and Group 4 (cryopreservation after 24-h cooling to 5°C and following CAM culture) showed that only preantral (primordial, primary, and secondary) follicles were viable. All the antral follicles in these three groups were degenerated (Fig. 2a) and, hence, were not counted.

Histological micrographs of follicles from ovarian tissue precooled to 5°C for 24 h and cultured in vitro for 8 days or cultured in the CAM system for 5 days. (a) Fresh (not cryopreserved) tissue after 24-h precooling to 5°C and 8 days in vitro culture. Degenerated antral follicles after cooling are shown by a bold black arrow. (b, c) Tissue after cryopreservation and 5-day CAM culture. (b) Tissue cryopreserved immediately after operation, without precooling. (c) Tissue cryopreserved after 24-h precooling to 5°C. (b1, b2) Degenerated follicles. (a1, b3, c1) Normal follicles. Scale bar: 25 μm.
The mean preantral follicle density per 1 mm3 was 12.9±2.8, 12.2±1.9, 12.4±3.3, 16.1±1.5 for Groups 1, 2, 3, and 4, respectively (p1–2 > 0.1; p3–4 < 0.05). It was detected that 87.0±1.6, 95.0±3.1, 71.0±3.9, and 84.0±1.2% follicles for Groups 1, 2, 3, and 4, respectively, were normal (p1–2, 3–4 < 0.05) (Fig. 3).

Effect of cooling on the quality of follicles [expressed as quantity (a) and percentage (b) of normal follicles]. Different superscripts indicate statistical differences between respective systems of culture, that is, in vitro versus in vitro (Group 1 vs. Group 2) and CAM versus CAM (Group 3 vs. Group 4) (p < 0.05; n = 8).
Immunohistology for Proliferation
Immunoreactivity of cells cultured without cooling varies from “lack of immunoreactivity” to “low,” whereas cells cultured with cooling varied from “low” to “high” (Figs. 4 and 5). The immunohistochemical analysis showed increased proliferation after cooling of fresh and cryopreserved tissue.

Cytokeratin expression in the cryopreserved and 5-day CAM-cultured tissue. (a–c) Three examples of tissue cryopreserved immediately after operation, without precooling. (d–f) Three examples of tissue cryopreserved after 24-h precooling to 5°C. Cytokeratin-positive cells are shown in red. Scale bar: 25 μm.

Ki-67 expression in the cryopreserved and 5-day CAM-cultured tissue. (a, a1) Tissue cryopreserved without precooling, immediately after operation. (b, b1) Tissue cryopreserved after precooling. Ki-67-positive cells are shown in brown. Scale bar: 25 μm.
Discussion
It is important to improve the posttransplantation survival of human ovarian tissue (1).
A positive effect of cooling of cells to low suprazero temperatures on their future development after rewarming has been observed before and is not new.
It was shown that the acclimation of the warm-blooded rat to cold stimulates mitosis indirectly in cells capable of division because it directly stimulates the mitotic activity in mouse and human cells cultured and adapted to the cold in vitro. In situ hybridization analysis of hypothalamic tissue showed that cold exposure causes a twofold increase in the total number of neurons expressing thyrotrophin-releasing hormone mRNA in the paraventricular nucleus (7).
In addition, our previous results showed good survival of bovine trophoblastic fragments that had been subjected to chilling for 48 h at 4°C. The survival/formation of vesicles in these fragments was not different from that of the untreated controls (both 98%) (14).
The exposure of human ovarian tissue to low positive temperatures of up to 26 h does not inhibit the development of follicles during subsequent in vitro culture. Compared with the untreated controls, the number of primordial follicles in all treatment groups significantly decreased owing to development to the advanced stages (10). The results of an investigation by Yin et al. (36), who found that, although ovaries showed fewer follicles, hypothermic storage of rat ovary at 4°C for 24 h did not disrupt ovarian function, does not support our results. This could be explained by the species-specific sensitivity of ovarian tissue to hypothermia.
Interesting investigations were performed by Wood et al. (35). The influence of long-term hypothermic storage of whole domestic cat ovary for 48 h at 4°C on follicleoocyte atresia and temporal taphonomy was investigated. It was found that the highest (but statistically insignificant) degeneration rate of follicles occurred at 48 h, with inhibition of taphonomy. Our data support these results.
Our experimental design included the use of two culture methodologies for ovarian tissue, in vitro, and CAM culture because animal testing has ethical and legal implications in Germany. Taking into account this fact as well as published results (13,21), we established chorioallantois membrane (CAM) of fertilized chicken eggs as a culture system in the present study. The chorioallantoic membrane system provides rapid results and may be used as a simple, inexpensive method in any laboratory. The extraembryonic vessel system of the chorioallantois membrane is similar to that of the mammalian placenta, which also is not innervated. This system is naturally immunodeficient. In addition, we used chick embryos in the first half of the usual breeding time up to the stage when the embryonic neural and immune systems are still underdeveloped. CAM can be used for xenotransplantation of different types of cells.
CAM culture of bovine and mice ovarian tissue has been reported by Cushman et al. (5) and Gigli et al. (8). It is established that primordial follicles during the CAM culture retained the ability to activate and grow. These authors believe that extracellular matrix constitution of CAM is similar to peritoneum, to which human ovarian tissue is transplanted by intraperitoneal xenotransplantation and orthotopic autotransplantation (5,8), CAM forms a bursa-like structure around ovarian pieces that is similar with in vivo condition. Recently, the similar investigations were performed on human ovarian tissue (13,21).
The CAM system is used to study angiogenesis in tumor (4) and endometriosis tissues (20,23). CAM also has been used for culture of human skin (17), liver (15), and skeletal muscle tissue (22), for surgical retinal research (18), for testing of different biomaterials in tissue engineering (32), and also for investigations of a bright spectrum on biological objects (30).
In conclusion, it appears that cooling of ovarian tissue to 5°C for 24 h before cryopreservation has a positive effect on viability of this tissue after thawing. Additionally, the efficacy of the CAM system for culture of thawed human ovarian tissue was demonstrated.
Footnotes
Acknowledgments
The authors are very grateful to Mr. F. V. Braun from University Bonn for helpful discussion of our results as well as to Ms. I. Orth and Ms. D. Peters for technical assistance. The authors declare no conflicts of interest.