
Abstract
Degeneration of midbrain dopamine neurons causes the striatal dopamine deficiency responsible for the hallmark motor symptoms of Parkinson's disease (PD). Intraparenchymal delivery of neurotrophic factors, such as glial cell line-derived neurotrophic factor (GDNF), is a possible future therapeutic approach. In animal PD models, GDNF can both ameliorate neurodegeneration and promote recovery of the dopamine system following a toxic insult. However, clinical studies have generated mixed results, and GDNF has not been efficacious in genetic animal models based on α-synuclein overexpression. We have tested the response to GDNF in a genetic mouse PD model with progressive degeneration of dopamine neurons caused by mitochondrial impairment. We find that GDNF, delivered to the striatum by either an adeno-associated virus or via miniosmotic pumps, partially alleviates the progressive motor symptoms without modifying the rate of neurodegeneration. These behavioral changes are accompanied by increased levels of dopamine in the midbrain, but not in striatum. At high levels, GDNF may instead reduce striatal dopamine levels. These results demonstrate the therapeutic potential of GDNF in a progressively impaired dopamine system.
Keywords
Introduction
Parkinson's disease (PD) is a common neurodegenerative disorder affecting about 1% of the population over the age of 60 (9) and is characterized by degeneration of dopamine (DA)-producing neurons in the substantia nigra pars compacta (SNc) and formation of intracellular inclusions called Lewy bodies. Although other populations of neurons are also affected, degeneration of SNc is responsible for the DA deficiency in the striatum that causes the disabling locomotor symptoms. Current treatments aim to alleviate symptoms by supplementing striatal dopamine levels or by other means restoring balance to affected basal ganglia circuitries, but there is currently no treatment that slows disease progression. Development of disease-modifying drugs has been hampered by an incomplete understanding of PD etiology as well as the pathophysiological events that lead to neurodegeneration. Two main hypotheses postulate toxicity from misfolded α-synuclein, the major component of Lewy bodies, and/or mitochondrial dysfunction (39). Major findings in support for a role for mitochondrial dysfunction include [1] PD patients have reduced mitochondrial respiratory chain function (33); [2] the toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), which causes DA neuron degeneration and Par-kinsonism in humans and laboratory animals, may exert its toxicity through inhibition of the respiratory chain (35); and [3] mitochondrial DNA (mtDNA) carrying large deletions, which accumulate in DA neurons during ageing and can cause respiratory chain impairment, are found in higher levels in SNc than in other brain regions, and in still higher levels in brains of PD patients (3,25).
Glial cell line-derived neurotrophic factor (GDNF) promotes the survival of dopamine neurons in vitro (27) and in vivo (40). Treatment with intraparenchymal GDNF, delivered by infusion, viral vectors, or transplantation of GDNF-producing cells, confers significant protection to DA neurons against 6-hydroxy dopamine (6-OHDA) or MPTP toxicity in rodent and primate PD models (1,6, 18,21,40). Clinical trials of GDNF delivery to PD patients have generated mixed results. Improved motor functions were found in two open-label studies (14,34), but a randomized control trial failed to reach its end point of motor symptom relief (24). Although the reason for this discrepancy is unknown, a concern is whether DA neurons affected by PD pathophysiology, such as α-synuclein accumulation and mitochondrial impairment, can respond to GDNF treatment (1,22). To this end, we have tested the efficacy of GDNF treatment in a mouse model with mitochondrial impairment in DA neurons.
We have previously developed a mouse model for PD by DA neuron-specific disruption of the gene encoding mitochondrial transcription factor A (Tfam) (10). Tfam is necessary for transcription and maintenance of mtDNA, which encodes essential subunits of the respiratory chain. These so-called MitoPark mice appear healthy at birth but suffer from progressive loss of striatal DA innervation and degeneration of DA neurons. Reduced spontaneous activity is first apparent from ~12 weeks of age and progressively decreases in parallel with striatal DA levels (11).
We now report that striatal delivery of GDNF can improve the motor phenotype of MitoPark mice but cannot modify the rate of neurodegeneration. Increased DA levels in the SNc, but not in the striatum, accompany these behavioral effects.
Materials and Methods
Construction, Packaging, Purification, and Titering of AAV Vectors
The construction of a self-complementing adeno-associated virus (AAV) vector expressing enhanced green fluorescent protein (AAV-GFP) has been described (44). A self-complementing AAV vector that expresses rat GDNF (AAV-GDNF) was made by replacing GFP with the cDNA for rat GDNF. Viral stocks of AAV-GDNF or AAV-GFP serotype 7 were prepared using a triple transfection method as described previously (17,45). Briefly, twenty 15-cm dishes (BD Falcon, Bedford, MA, USA) containing HEK293 cells (American Type Culture Collection [ATCC], Manassas, VA, USA) at 85–95% confluence were transfected by the CaCl2 method (with pHelper, Stratagene, La Jolla, CA, USA), pdsAAV-GDNF (5), or pdsAAV-GFP (44) and pAAV7 (31). The vectors pdsAAV-GFP, pdsAAV-GDNF, and pAAV7 were generously provided by Dr. Xiao Xiao, University of North Carolina (Chapel Hill, NC, USA). pXR5 was obtained through the National Gene Vector Biorepository (Indiana University School of Medicine, Indianapolis, IN, USA). Approximately 48 h post-transfection, cells were harvested, lysed by freezing and thawing, and purified by centrifugation on a CsCl (Sigma-Aldrich, St. Louis, MO, USA) gradient. Final samples were dialyzed in PBS (Sigma), aliquoted, and stored at −80°C. The amount of viral genomes (vg)/μl was determined by quantitative PCR of the cytomegalovirus promoter.
In Vitro GDNF Measurements
Primary cortical cultures were prepared and transduced on DIV8 as described previously (17). Cells were plated in 24-well plates (800 μl media per well; Costar, Corning, Corning, NY, USA) and transduced with 50 μl of concentrated, 1:5 or 1:10 diluted virus. On DIV11 at 72 h posttransduction, media were collected for GDNF ELISA. GDNF levels in media were measured using a commercial immunoassay (EMax Immunoassay kit, Promega, Madison, WI, USA).
Animal Breeding
Dopamine transporter cre recombinase-expressing (DATcre; generated in house) (10) and loxP-flanked Tfam (TfamloxP; kindly donated by Prof. N. G. Larsson, Karolinska Institutet, Stockholm, Sweden) (26) strains were back-crossed at least 10 generations to a C57Bl/6 background. To generate MitoPark mice, DAT+/cre and TfamloxP/loxP mice were first intercrossed to obtain double heterozygotes. Double heterozygote males (DAT+/cre; Tfam+/loxP) were then crossed to homozygous TfamloxP/loxP females to generate MitoPark mice (genotype DAT+/cre; TfamloxP/loxP) and littermate controls (genotype DAT+/+; TfamloxP/loxP or DAT+/+; Tfam+/loxP). Experiments were approved by an ethical committee and performed in compliance with Swedish law.
In Situ Hybridization
GDNF transcript was detected using a radiolabeled oligonucleotide probe (5′-GGCCGCTTCACAGGAACCGCT ACAATATCGAAAGATCAGTTCCTCCTTGG) based on a published protocol (7). The probe was end-labeled with α-33P-deoxyadenosine 5′-triphosphate using a terminal deoxynucleotidyl transferase kit (GE Healthcare, Uppsala, Sweden). Coronal cryosections of striatum from freshfrozen brains were hybridized overnight at 42°C with radiolabeled probe in 4×SSC solution (3.0 M trisodium citrate and 3.0 M NaCl; pH 7.0; both from Merck KGaA, Darmstadt, Germany), 50% formamide, 1× Denhardt's solution (Ficoll, polyvinylpyrrolidone, and bovine serum albumin), 1% sarcosyl, 0.02 M Na3PO4 (Merck), 10% (w/v) dextran sulfate, 0.2 M dithiothreitol (DTT), and 0.5 μg/μl sheared salmon serum sperm DNA (all from Sigma unless specified otherwise). Following five washes in 60°C 1×SSC buffer, slides were dehydrated in an ethanol series and air-dried. Signal was detected by exposure to autoradiographic film (BioMax, Eastman Kodak, Rochester, NY, USA).
Immunohistochemistry and Cell Counts
Mice were deeply anesthetized with pentobarbital and perfused with heparinized Ca2+-/Mg2+-free Tyrode's solution (137 mM NaCl, 2.7 mM KCl, 0.2 mM Na2HPO4, 12 mM NaHCO3, 11 mM d-glucose; pH 7.6; all from Sigma) followed by fixative (4% paraformaldehyde with 0.4% picric acid in 0.16 M phosphate buffer; all from Merck). Brains were dissected, postfixed, and subsequently equilibrated with 10% sucrose (Merck) in phosphate buffer. Brains were frozen on dry ice and cryosectioned.
Coronal brain sections were immunolabeled using antibodies against tyrosine hydroxylase (TH; Pel-Freez, Rodgers, AR, USA; 1:500 dilution, overnight at 4°C) and DAT (DAT-Nt, Millipore, Solna, Sweden; 1:200 dilution) and detected by a biotinylated secondary antibody (Vector Laboratories, Peterborough, UK; 1:200 dilution, 1 h at room temperature) and a peroxidase substrate (Vector SG, Vector Laboratories). Images were acquired using a Zeiss axiophot 2 microscope equipped with 5×/0.45NA, 20×/0.6NA, and 40×/0.65NA objectives and Axioplan 2 Imaging Software (Zeiss, Stockholm, Sweden). SNc was outlined as described (2), and every sixth section was used for counting. Nuclei of TH-positive neurons in SNc were counted using a 20× objective and with the genotype and treatment of the subjects blinded to the investigator. For Abercrombie's correction, nuclear diameter was measured as maximal diameter in the x/y plane of randomly selected TH-labeled cells in SNc (n≥25 per subject).
Spread of GFP and TH density in coronal striatal sections (14 μm) was assessed using confocal microscopy (LSM510, Zeiss). TH was labeled using cyanine 3 (Cy3)-conjugated secondary antibodies (1:400 dilution; Jackson Biolabs, Bar Harbor, ME, USA). TH density was measured in subsampled areas of striatum (326×326 μm; z <1.0 μm; 6 per section, 3 sections per brain) excluding the injection tracts. A 40×/1.3NA oil objective was used to detect Cy3 excited at 543 nm and detected at 553-649 nm. Image area occupied by TH-immunoreactive fibers was measured using ImageJ software (NIH, Bethesda, MD, USA). GFP was excited with a 488-nm laser and detected at 499-510 nm.
Animal Genotyping
For genotyping, ear skin biopsies were boiled (98°C) for 45 min in 100 μl 1× lysis buffer (10×: 250 mM NaOH, 2 mM EDTA; both from Merck) and then cooled on ice and mixed with 100 μl 1× neutralizing buffer (10×: 400 mM Tris-Hcl, pH 7.5; Merck). One microliter of the solution was used for a 25-μl PCR reaction with 1 unit Taq polymerase (DyNAzymeII, Finnzyme, Espoo, Finland) and 0.2 μM of each primer. For genotyping the DAT locus, primers 3F (5′-CATGGAATTTCAGGTGCTTGG), 3R1 (5′-ATGAG GGTGGAGTTGGTCAG), and 3R2 (5′-CGCGAACATCT TCAGGTTCT) were used for amplification [95°C, 10 min; 38× (95°C 30 s, 58°C 30 s, 72°C 35 s); 72°C 5 min]. This generates a ~310-bp product for the wild-type allele and a ~470-bp product for the DATcre allele. For Tfam, primers F1 (5′-CTGCCTTCCTCTAGCCCGGG), R1 (5′-GTAAC AGCAGACAACTTGTG), and R2 (5′-CTCTGAAGCA CATGGTCAAT) amplify [95°C, 10 min; 42× (95°C 30 s, 58°C 30 s, 72°C 45 s); 72°C 5 min] a 404-bp band for the wild-type allele and a 437-bp band for the TfamloxP allele. PCR products were separated on 1.5% or 4% agarose gels (Sigma), respectively.
Stereotaxic Surgery and Viral Delivery
Mice were anesthetized with isofluorane (Baxter, Kista, Sweden) and buprenorphine (RB Pharmaceuticals Ltd.; Stockholm, Sweden; 0.075 mg/kg SC), and placed in a stereotaxic frame (Stoelting, Dublin, Ireland). A midline skin incision was made, and bregma was identified. Following penetration of the skull bone using a small dental drill (Desk 300IN, Silfradent, Sofia, Italy), a 10-μl Hamilton syringe (Reno, NV, USA) with a custom-made 33-G needle (45° type 4 tip) was lowered into striatum bilaterally (0.2 mm anterior to bregma, 1.8 mm lateral to the midline, and 3.8 mm ventral to dura mater). A microsyringe pump controller (World Precision Instruments, Hitchin, Hertfordshire, UK) was used to deliver 1 μl of viral suspension at a speed of 5 nl/s and another 1 μl following 0.3 mm retraction of the needle (to 3.5 mm ventral to dura mater), after which the needle was left in situ for another 4 min before retraction. Each side thus received a total of 2 μl (1×109 viral genomes). A 6-0 Ethicon suture (Johnson & Johnson, Sollentuna, Sweden) was used to close the scalp.
Protein Delivery Via Osmotic Minipumps
Osmotic minipumps (Alzet 1004, Durect Corporation, Cupertino, CA, USA) were primed in saline and filled with recombinant GDNF (a kind gift from Don Gash, University of Kentucky, Lexington, Ky, USA) or vehicle (saline). Via stereotaxic surgery (described above), a brain infusion cannula (Durect, Cupertino, CA, USA) was implanted in the right striatum (0.2 mm anterior to bregma, 1.8 mm lateral to the midline, and 3.8 mm ventral to dura mater). The cannula was anchored to the skull bone using dental cement (Dentalon, Tel Aviv, Israel). Polyethylene tubing (PE50, VWR, Stockholm, Sweden) connected the cannula to the pump, which was implanted underneath the back skin. Pumps delivered 0.5 μg GDNF per 24 h during 4 weeks. Mice with apparent pump failure were omitted from analysis.
Behavior Tests
Spontaneous behavior was measured between 8 and 12 AM during the light phase of the light/dark cycle. Mice were habituated to the dimly lit, low-noise, and ventilated room for at least 30 min before initiating experiments. Mice were individually placed in activity boxes (40×40 cm; VersaMax, AccuScan Instruments, Columbus, OH, USA) and a grid of infrared light beams at floor level and 7.5 cm above recorded horizontal and vertical locomotor activities, respectively. Measurements were acquired per 5-min bin over a period of 60 min.
Measurements of Monoamines
DA and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were measured by high-performance liquid chromatography (HPLC) as previously described (36). Briefly, mice were sacrificed by cervical dislocation, and brains were rapidly removed and chilled in ice-cold saline. Bilateral pieces of striatum, midbrain, and frontal cortex were dissected and frozen at −70°C until analysis. Tissue pieces were homogenized by sonication in 0.1 M perchloric acid (Merck) containing 0.4 mM sodium bisulfite (Sigma). DA, HVA, and DOPAC in the filtered supernatant were separated on a reversephase column and measured by electrochemical detection (Coulochem III, ESA, Chelmsford, MA, USA).
Statistical Tests
Data are represented as mean ± SEM. Statistical testing of the effects of GDNF treatment was carried out using unpaired Student's two-tailed t tests (following a Levene's test for equality of variance), paired two-tailed t tests, univariate ANOVA with Fisher's LSD post hoc tests, or univariate general linear models (GLM) using appropriate software (SPSS 20; IBM, Armonk, NY, USA). Differences between control and MitoPark mice are expected and thus not indicated. Values of p < 0.05 were considered significant.
Results
Long-term trophic support could allow modification of the slow and progressive degeneration of DA neurons in PD and in MitoPark mice. Gene delivery by AAV has shown beneficial effects in rodent PD models (19,29,30,43). We packaged the GDNF cDNA into AAV of serotype 7, which primarily transduces neurons and results in widespread expression in striatum (38). To measure GDNF production from AAV-GDNF, rat primary cortical neurons (at DIV8) were transduced with different concentrations of AAV-GFP or AAV-GDNF. We assessed the levels of GDNF secreted in the media 72 h posttransduction by an ELISA assay. AAV-GFP did not increase GDNF levels above background whereas AAV-GDNF produced 1,410 ± 150 pg GDNF/109 vector genome (vg) (or 1 μl virus) over 72 h (Fig. 1A).

GDNF synthesis by AAV7 and spread in striatum. (A) Rat primary cortical neurons (DIV8) were transduced with various amounts of adeno-associated virus-green fluorescent protein [AAV-GFP; 4×109 vector genome (vg)/μl] or AAV-glial cell line derived growth factor (AAV-GDNF; 1×109 vg/μl). GDNF protein was significantly elevated in AAV-GDNF-treated cultures. (B) Distribution of GFP-fluorescent neurons along the injection tract in striatum 3 weeks following a single injection of AAV-GFP (3×10 10 vg). Scale bars: 500 μm (overview) and 50 μm (magnification). (C) Midbrain section showing absence of transduced DA neurons in substantia nigra pars compacta (SNpc) but GFP-positive fibers in substantia nigra pars reticulata (SNr). Scale bar: 500 μm. (D) GDNF expression 3 weeks after intrastriatal delivery of 1×10 9 vg of AAV-GDNF and -GFP, as determined by in situ hybridization with an oligonucleotide complimentary to GDNF. The signal was sensitive to RNase treatment and not found using a negative control (random) probe. Approximate distances relative to bregma are indicated.
Intrastriatal delivery of AAV-GFP resulted in widespread GFP expression in striatal neurons (Fig. 1B). Some GFP-expressing cells lining the ventricles in the corpus callosum and in the hippocampal formation were also seen. At the level of the midbrain, viral uptake in SNc was not detected, but substantia nigra pars reticulata (SNr) contained numerous GFP-positive fibers (Fig. 1C). In situ hybridization revealed widespread localization of GDNF mRNA in striatum on the injected side (Fig. 1D).
We studied the effects of GDNF overexpression in the MitoPark model by bilateral striatal injections of AAV in young (6–7.5 weeks) MitoPark and control mice. At this age, MitoPark mice still have normal striatal dopamine levels and show no symptoms of parkinsonism (11). Each mouse was randomized to receive either AAV-GDNF (1×109 vg/side) or equal titers of AAV-GFP. Following surgery, we used locomotor activity boxes to follow spontaneous locomotion (horizontal) and rearing (vertical) activities (Fig. 2A). AAV-GDNF produced no significant changes in locomotion or rearing activity during the first 4 weeks after injections (Fig. 2B). Eight weeks after surgery, when mice were ~15 weeks old and motor symptoms in MitoPark mice normally appear, treatment with AAV-GDNF significantly increased locomotor performance (+31% compared with MitoPark receiving AAV-GFP, p = 0.003) (Fig. 2B, C). Control mice receiving GDNF also showed a small increase in locomotor activity (+17%, p = 0.048) at this time point. Rearing activity, which is more sensitive to DA function and decreases prior to locomotion, improved significantly (+99% compared with GFP, p = 0.003) in MitoPark mice receiving GDNF (Fig. 2B, C). GDNF can thus partially attenuate the progressive locomotor dysfunction in MitoPark mice.
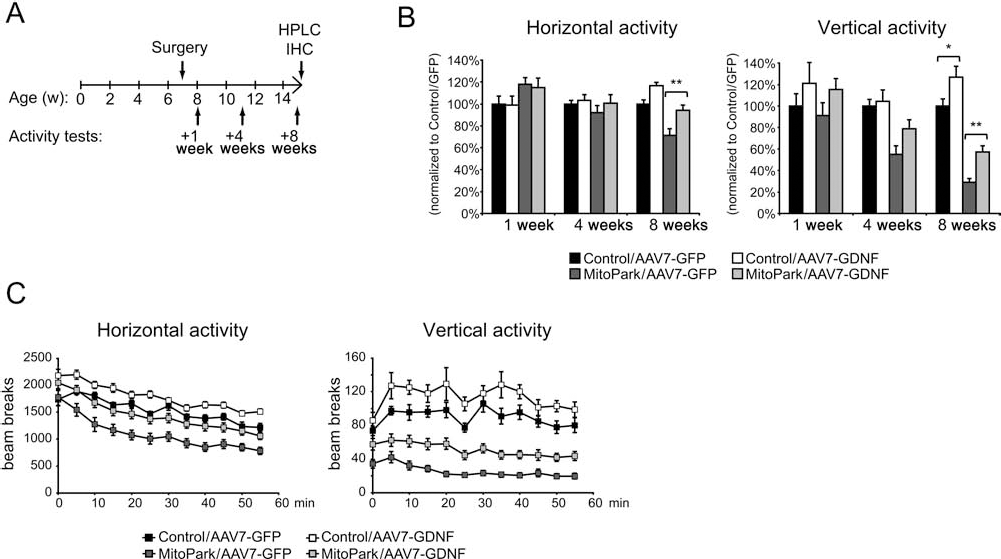
AAV-GDNF improves locomotor phenotype in MitoPark mice. (A) Illustration of experimental setup. (B) Spontaneous horizontal activity (locomotion) and vertical activity (rearing) 1, 4, and 8 weeks after AAV injections. Values are normalized to the means of controls receiving GFP. Data shown as mean ± SEM (n = 8–12, except for MitoPark/8 weeks groups, which were expanded to n = 20–23 by addition of balanced cohorts). *p < 0.05, **p < 0.01 by ANOVA. (C) Horizontal activity (locomotion) and vertical activity (rearing) plotted over time for groups assessed 8 weeks after AAV delivery. HPLC, high-performance liquid chromatography; IHC, immunohistochemistry.
To investigate whether GDNF affected the rate of neurodegeneration in MitoPark mice, we studied coronal sections of the midbrain DA system immunolabeled with an antibody against tyrosine hydroxylase (TH), a marker for DA neurons. TH-positive neurons of GDNF-treated brains looked similar to those of GFP-treated mice of the same genotype (Fig. 3A) but showed a tendency towards increased soma perimeter length (+8%, p = 0.07). We counted TH-positive neurons in SNc 8 weeks after striatal AAV injections (Fig. 3B) and found that MitoPark mice at this age (~15 weeks) had a ~40% reduction in cell numbers, consistent with previous reports (10). There was no difference between MitoPark mice receiving GDNF compared to those receiving GFP. In striatum, no increase in dopaminergic innervation was seen in GDNF-treated mice, using antibodies against both TH and DAT (Fig. 3C). We measured the density of TH-positive fibers in thin optical sections of randomly chosen striatal areas (Fig. 3D). MitoPark mice had ~80% reduction compared to control mice, consistent with previous data (37), and those receiving GDNF, a nonsignificant lesser TH-positive fiber density than those receiving GFP (p = 0.07; ANOVA). We conclude that trophic support by GDNF cannot slow down the progressive degeneration of MitoPark DA neurons and fails to induce significant compensatory sprouting.

GDNF treatment does not modify rate of neurodegeneration in MitoPark mice. (A) Tyrosine hydroxylase (TH)-positive dopamine (DA) neurons in SNc of a control and a MitoPark mouse 8 weeks following striatal delivery of AAV-GFP or AAV-GDNF. (B) Number of TH-positive neurons in SNc of control or MitoPark mice 8 weeks following striatal delivery of AAV-GFP or AAV-GDNF. Absolute numbers were calculated using Abercrombie's formula and found to be (mean ± SEM) 6,634 ± 551 in the control/GFP group (n = 3), 7,206 ± 208 in the control/GDNF group (n = 3), 4,107 ± 372 in the MitoPark/GFP group (n = 5), and 4,110 ± 186 in the MitoPark/GDNF group (n = 5), based on nuclear diameters 9.75 ± 0.15, 9.87 ± 0.46, 9.25 ± 0.25, and 9.66 ± 0.29 μm, respectively. (C) TH- and dopamine transporter (DAT)-reactive fibers in striatum. Scale bars: 500 μm and 20 μm. (D) Quantification of TH-reactive fibers in striatum, determined by the area fraction occupied by labeled fibers in confocal micrographs. Values obtained for control/GDNF (n = 2), MitoPark/GFP (n = 5), and MitoPark/GDNF (n = 5) were normalized to that of the control/GFP group (n = 2).
Next, we measured the levels of dopamine and its major metabolites in brain homogenates by HPLC. At the level of substantia nigra, GDNF increased DA levels in both control (+62% compared with GFP, p = 0.046) and MitoPark (+107% compared with GFP, p < 0.001) mice (Fig. 4A). In parallel, major DA metabolites DOPAC and HVA also increased (Fig. 4A). In striatum of control mice, GDNF had no affect on DA levels (Fig. 4B) but increased the amounts of DA metabolites DOPAC (+28%, p = 0.04) and HVA (+30%, p < 0.001). Increased metabolite/DA ratios have previously been seen following striatal GDNF delivery and may reflect increased DA turnover (13,16). However, a similar response was not seen in MitoPark mice, and GDNF also had no effect on striatal DA levels in MitoPark mice (Fig. 4B).

Improved dopamine homeostasis in SNc but not striatum of GDNF-treated mice. (A) Levels of dopamine, dopamine metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) and metabolite/DA ratios in substantia nigra of control (n = 6–7) and MitoPark (n = 18) animals treated with AAV-GFP or AAV-GDNF, respectively. (B) Dopamine, metabolites and metabolite/DA ratios in striatum. *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA.
The above data were corroborated in a separate and smaller study, in which osmotic minipumps were used for chronic unilateral delivery of recombinant GDNF protein or vehicle into striatum (Fig. 5A). Pumps were primed to deliver a high dose of GDNF (0.5 μg/day) continuously over 4 weeks and were implanted in 12-week-old MitoPark mice (Fig. 5B), which were studied during the following 6 weeks. Mice receiving GDNF showed significant improvement in spontaneous locomotion (horizontal) activity 6 weeks after treatment (Fig. 5C). We measured DA levels in SNc and striatum of the mice 6 weeks after pump implantation. SNc homogenates ipsilateral to the injection site contained significantly higher levels of DA than the contralateral (uninjected) side. This was true for both GDNF and vehicle delivery but was much more pronounced following GDNF treatment. DA levels in ipsilateral SNc of GDNF-treated mice tended to be higher than the DA levels in mice receiving vehicle (+99%, p = 0.08). In analogy with previous results, striatal DA levels did not increase. Instead, mice receiving GDNF had reduced striatal DA levels (-57%, p < 0.05) compared to the control group. This may reflect a downregulation of TH in striatum (32), which has also been observed following long-term lentiviral delivery of GNDF into denervated striatum (12).

Effects of striatal delivery of recombinant GDNF protein to MitoPark mice mimic the effects of gene delivery. (A) Spread of GDNF in striatum after 3 weeks of continuous delivery to unilateral striatum. (B) Illustration of experimental setup. (C) Spontaneous horizontal activity (locomotion) and vertical activity (rearing) of MitoPark mice 2, 4, and 6 weeks after pump implantation. *p < 0.05 and ‡p < 0.1 by two-tailed t tests. (D) Dopamine levels in unilateral substantia nigra (n = 5–8). *p < 0.05, **p < 0.01 by paired two-tailed t tests, comparing the injected and non-injected sides of each animal. ‡p < 0.1 by univariate GLM, comparing injected sides. (E) Dopamine levels in unilateral striatum. *p < 0.05 by univariate GLM, comparing injected sides.
Our data thus show that increased expression of GDNF can partially alleviate motor symptoms in MitoPark mice, possibly by increasing DA levels in the substantia nigra area. However, GDNF is unable to slow the progressive loss of DA neurons in MitoPark mice. At high levels, it may instead accelerate loss of striatal TH reactivity and DA levels.
Discussion
In experimental PD models, GDNF signaling confers increased resistance to toxins such as 6-OHDA and MPTP and also promotes recovery of the nigrostriatal DA system following such insult. Mice lesioned with MPTP can slowly recover nigrostriatal innervation by mechanisms that involve GDNF-Ret (rearranged during transfection) signaling (23). In PD patients, GDNF stimulates DA turnover and may alleviate motor symptoms, but it is not known whether such treatment has an effect on cell survival. Unlike toxin-based animal models, PD is a progressive disorder that likely impacts DA neurons well before overt degeneration. Such impaired neurons may have lower regenerative capacity than those in most PD models, in which neurons recover from an acute or subacute toxic insult. Indeed, the relevance of the toxin-based models has been challenged by two different studies, in which GDNF failed to confer protection against α-synuclein toxicity mediated by viral overexpression (8,28). Somewhat akin to the presumed situation in PD, DA neurons are functionally impaired prior to overt behavioral symptoms in MitoPark mice. For example, DA release and DA neuron pacemaker activity, as well as hyperpolarization-activated cyclic nucleotide-gated (HCN) ion channel function, are impaired at 6–8 weeks of age in these mice (15). Moreover, MitoPark DA neurons have an impaired anterograde supply of mitochondria to distal axons (37). Such DA neuron dysfunction is compatible with our finding that GDNF was unable both to induce sprouting of TH-positive nerve terminals and to delay degeneration of DA neurons caused by mitochondrial failure in MitoPark mice.
GDNF in striatum is taken up by DA neurons and transported in retrograde direction to the cell bodies in the SNc (41). MitoPark neurons have retained but decreased retrograde transport (37), which may reduce bioavailability of GDNF in SNc. Nevertheless, we found positive effects on animal behavior and midbrain DA levels, showing that GDNF may relieve PD motor symptoms without being neuroprotective. However, the behavioral effects were not associated with increased striatal DA levels. Although the explanation for this is unknown, a similar discrepancy has been found in other studies (4,16,42). It has been suggested that GDNF can partly restore basal ganglia function by modifying signaling of dendrodendritic interconnections between SNc and SNr (42). The relatively decreased striatal DA levels may, at least in part, result from a combination of GDNF-induced stimulation of DA release (20) with opposing processes such as dysfunctional DA synthesis or reuptake. Impaired anterograde axonal transport (of mitochondria and possibly other cellular components) may also leave the distant terminals unresponsive to trophic effects of local GDNF, while at the same time sufficient amounts of GDNF protein undergo retrograde transport to exert effects at cell body and dendrite levels.
Our findings with bilateral delivery of the GDNF gene to striatum in MitoPark mice were supported by our results with direct delivery of GDNF protein. While such delivery, using osmotic minipumps, was unilateral for technical reasons, we still noted improvements of behavior and increases of DA in the DA cell body area, but not in striatum. These observations are in agreement with the findings using viral GDNF gene delivery and thus support the general conclusion that GDNF delivery to striatum leads to significant motor improvements without rescue of TH-positive fibers or increased striatal DA levels. Instead, reduced striatum DA levels were found following long-term delivery of recombinant GDNF, while mesencephalic DA levels, at the same time, increased. High lentiviral-mediated expression of GDNF has previously been found to also downregulate TH in the intact DA system (13,32), but divergent results have been obtained regarding the consequences for striatal DA levels. We did not observe any loss of TH immunoreactivity in control mice following AAV-GDNF delivery, possibly due to lower levels of expression with AAV. A tendency towards reduced TH reactivity in striatum was however observed in MitoPark mice receiving AAV-GDNF.
We conclude from our model that DA neurons with severe impairment of respiratory chain function may still moderately benefit from exogenous GDNF. The MitoPark model represents a severe form of mitochondrial dysfunction, and it is possible that DA neurons in PD have less severe dysfunction and are more responsive to neurotrophic treatment. However, our data also suggest that long-term delivery of high GDNF doses into striatum may have negative effects, such as decreasing local DA levels. This underscores the importance of careful dosage titrations and safety assessments in clinical trials of neurotrophic factor delivery for PD.
Footnotes
Acknowledgments
This work was supported by the National Institutes of Health-Karolinska Institute Graduate Partnership Program for the Neurosciences (F.H.S.), the Swedish Research Council, Swedish Brain Power, the Parkinson Foundation, the Karolinska Institutet, and US Public Health Service (National Institute on Drug Abuse, Intramural Research Program and R01NS070825). L.O. is a shareholder in a company that holds commercial rights to MitoPark mice.