
Abstract
Cord blood is regarded as a powerful source for adult stem cells. Cord blood transplants have been used successfully to treat children and adults in autologous and allogeneic settings. Nevertheless, in many cases, the clinically relevant cell number (CD34+ cells and total leukocytes) is a limiting factor. To enable standardized cell banking and future in vitro expansion of adult stem/progenitor cells, elimination of serum, which inevitably differs from lot to lot and donor to donor, is highly desirable. Here, we demonstrate the feasibility of a xeno-free, chemically defined cryopreservation procedure for cord blood-derived cells over a period of 1 year. Cell recoveries with respect to retrieval of clinically relevant CD34+ cells, colony-forming units, and in vitro cultures of erythroid progenitor cells under standardized conditions were analyzed after 1 week or 1 year of cryopreservation and found to be very high and similar to the samples before freezing. The established xeno-free procedure is an important step toward using the full potential of adult stem cells from cord blood, enabling the elimination of serum-derived factors negatively influencing proliferation, differentiation, and survival of hematopoietic stem cells.
Keywords
Introduction
The allogeneic transplantation of umbilical cord blood is widely accepted as a therapeutic treatment of hematological disorders and malignant diseases in children and adults (2, 6, 9–11, 13, 21, 22). Umbilical cord blood transplantation (UCBT) has several advantages to bone marrow transplantation (BMT) (15): (i) the fast procurement due to thousands of stored and human leukocyte antigen (HLA) typed cord blood units worldwide; (ii) lower incidence of severe graft-versus-host disease (GVHD); and (iii) less stringent requirements for HLA matching. However, there are also major disadvantages: (i) delayed engraftment and increased risk of graft failure compared to BMT; (ii) delayed T-cell immune reconstitution; and (iii) increased costs of hospitalization. Due to the continued shortage of HLA-matched bone marrow donors, UCBT is successfully used to treat a broad variety of malignant and nonmalignant diseases when no HLA-matched BMT is available. An adequate supply of high-quality cord blood cells can only be guaranteed upon establishing standardized and efficient protocols for cryopreservation and storage (8). Nowadays, most cord blood banks store red cell-depleted, volume-reduced samples (leukocyte-rich plasma, LRP) that are cryopreserved in a cocktail of dimethyl sulfoxide (DMSO) and dextran-40 (8, 23). The stored products are thawed and subsequently washed in order to reduce the DMSO concentration before infusion. Total leukocyte recoveries after cryopreservation typically are close to 80%, which means that a substantial amount of dead/apoptotic cells is transfused into the recipient. Therapy success, however, always correlates with the transfused cell dose (total CD34+ cells) (6, 21, 22). Recently, several cases of cardiovascular toxicity have been reported (12, 16), and Ma et al. argued that dextran-40 could be responsible for this adverse effect (16). These reports indicate that the cryopreservation of cord blood cells should be improved to maximize the number of transfused cells and to avoid transplant-induced complications in the diseased recipients. One solution for the persistent problem of inadequate cell numbers in cord blood transplants could be the in vitro expansion of hematopoietic stem cells (HSCs) and progenitor cells. Successful ex vivo expansion of HSCs has already been demonstrated in the mouse system (19, 30). This approach has already been envisaged in the year 1996 as “the next generation of cellular therapeutics” (7), but its transformation to the clinic has proven to be difficult. To enable standardized expansion protocols for cord blood cells, it is important to avoid human plasma, which is contained in most stored cord blood samples. Cytokines like tumor necrosis factor-α (TNF-α) or transforming growth factor-β (TGF-β) in the plasma may negatively influence in vitro cell expansion even at very low concentrations. Therefore, a fully defined cryopreservation medium would facilitate standardized conditions for subsequent cell expansion by avoiding additional cytokines contained in most cryomedia. Cells derived from cord blood are generally not used directly for therapy, but have to be stored for several years, demanding high standards for the cryopreservation protocol and the storage conditions. Efficient recovery of hematopoietic progenitor cells after long-term cryopreservation has been reported (4). Recently, long-term cryopreserved cord blood cells have been used for competitive repopulation experiments in mice, thereby demonstrating that true HSCs are still functional after more than 20 years of cryopreservation (3). In addition, the same group also used the cryopreserved cord blood as a starting material for reprogramming into induced pluripotent stem (iPS) cells and for the isolation of endothelial progenitors, demonstrating the therapeutic potential of the stored cord blood units (3). In a recent study, we have characterized a xeno-free, fully defined cryomedium, which gave excellent recoveries of different stem/progenitor cell types after cryopreservation (29). This cryomedium, which is chemically fully defined, was tested with three different primary cell types: umbilical cord blood (UCB)-derived erythroid progenitor cells (EPCs), UCB-derived endothelial colony-forming cells, and adipose tissue-derived mesenchymal stromal cells. For all three cell types, cell recovery after cryopreservation was equal to or better than a reference medium consisting of 90% fetal bovine serum (FBS) and 10% DMSO (29).
The aim of the present study was to evaluate the long-term storage of cord blood cells in this xeno-free and chemically fully defined cryomedium. The efficiency of the cryopreservation procedure was assessed by measuring the viability and recovery of total mononuclear cells (MNCs) and CD34+ cells, as well as the amount of colony-forming units (CFUs). Standardized EPC cultures were performed to characterize the potential for in vitro expansion cultures. EPC cultures were chosen as a commonly used and well-characterized model for expansion cultures from cord blood because of their high proliferative potential, and the ability to perform cultures from small aliquots of cryopreserved cord blood cells, which was a prerequisite for our study.
Materials and Methods
Cord Blood Collection
Cord blood samples were collected after obtaining informed consent from the mother according to the institutional guidelines of the General Hospital of Vienna. Cord blood was collected by venipuncture after cesarean section or spontaneous birth into 250-ml cord blood bags containing 35 ml of citrate phosphate dextrose adenine (CPDA-1) anticoagulant solution (Baxter Healthcare, Vienna, Austria). Samples with cord blood volumes below 50 ml were excluded. The collected cord blood was stored at room temperature until the isolation of the mononuclear cells (MNCs) was started. Isolation of MNCs, determination of cell viability, quantification of CD34+ cells, establishing prefreezing cultures and CFU assays, and freezing of cells were completed within 12 h after isolation of the cord blood.
Isolation of MNCs From Cord Blood
The cord blood was dispensed into 50-ml Falcon tubes (BD Biosciences, Franklin Lakes, NJ, USA) under sterile conditions and diluted 1:2 with Dulbecco's PBS (Gibco/Invitrogen, Carlsbad, CA, USA). The diluted cord blood was carefully layered on top of a Ficoll cushion (Biocoll Separating Solution, Biochrom, Berlin, Germany). The tubes were centrifuged at room temperature (RT) for 40 min at 600 × g. Next, the blood/Ficoll interphase was collected, diluted with PBS, and the cells were pelleted (300 × g, 10 min, RT) and resuspended in 50 ml of hypotonic lysis buffer (8.99 g of ammonium chloride, 1 g of KHCO3, 0.037 g of EDTA per liter, pH to 7.3; all from Sigma-Aldrich, St. Louis, MO, USA). Aliquots were removed for cell counting (CASY counter; Roche-Innovatis, Indianapolis, IN, USA) and determination of CD34+ cells. The MNCs were then resuspended in Fraunhofer Institute for Biomedical Engineering (IBMT) medium (available from Fischer Procryotect, Ruedlingen, Switzerland) at a concentration of 1 × 107 cells/ml. The medium contains a protein-free basal medium, an ethylene oxide/propylene oxide block copolymer (Pluronic F-68), and DMSO as cryoprotectant, and does not contain any additional proteins. For cell viability determination in triplicates and CD34 analytics, one aliquot was further diluted 1:10 with cryomedium and dispensed into 10 aliquots containing 1 × 106 cells/ml. For prefreeze analysis, one aliquot of 1 × 107 cells and three aliquots of 1 × 106 cells were processed for CFU assays and in vitro cultivation and cell viability analysis, respectively. The other samples were frozen as outlined in Figure 1 and in the section below.
Freezing and Thawing of Cells
Isolated mononuclear cells were placed on ice and transferred to a temperature-controlled freezer (SY-LAB, Purkersdorf, Austria). The cells were frozen with the following protocol: 4°C for 30 min (precooling), temperature gradient −1°C/min from 4°C to −80°C, −80°C for 2 h (hold). Frozen cells were transferred to the gas phase of a N2 storage tank (Taylor-Wharton K series equipped with CryoCon AFT-3L module for temperature monitoring and automatic N2 refill; Taylor-Wharton Cryogenics, Theodore, AL, USA) and stored for the indicated periods. For thawing, cells were transferred from the storage tank to a 37°C water bath and swirled continuously until thawed. The cryovials were immersed in 70% EtOH, dried, and transferred to a laminar flow cell culture hood. After thawing, the cells were washed once with 10 ml of prewarmed Roswell Park Memorial Institute (RPMI) medium (Lonza, Basel, Switzerland), pelleted (300 × g, 5 min, RT), and resuspended in StemSpan medium (STEMCELL Technologies, Vancouver, Canada) for cultivation.
Cell Viability Analysis with the 1 × 106 MNC Aliquots
To enumerate the viable cells, triplicate samples from each cord blood sample were analyzed at each time point (prefreeze, 1 week cryo and 1 year cryo). Two hundred microliters of the cell suspension was transferred to fluorescence-activated cell sorting (FACS) tubes (BD Biosciences) and diluted with 300 μl of PBS. Dead cells were stained by addition of 5 μl of propidium iodide (PI) staining solution [50 μg PI/ml in PBS (pH 7.4)], and the fraction of viable cells (PI negative) was determined by flow cytometry on a FACScan (BD Biosciences). Nucleated cell counts were determined with the CASY counter (60-μm capillary, size range 5–15 μm). The number of viable MNCs was calculated by multiplication of the percentage of viable cells with the total number.
Cultivation of EPCs From 1 × 107 MNC Aliquots
The mononuclear cells resuspended in cryomedium were washed once with 10 ml of RPMI medium and pelleted (300 × g, 5 min, RT), and the supernatant was aspirated carefully. The cell pellets were resuspended in 1 ml of StemSpan medium (STEMCELL Technologies, Vancouver, Canada), and an aliquot was removed for the CFU assays. Then, the cells were supplemented with 2 U/ml erythropoietin (Erypo, 10,000 U/ml, Jannsen-Cilag, Vienna, Austria), 100 ng/ml stem cell factor (SCF), 1 × 10−6 M dexamethasone (Dex), 40 ng/ml insulin-like growth factor-1 (IGF-1), and 20 μg/ml of a cholesterol-rich lipid mix (all from Sigma-Aldrich) and cultivated as described (5, 14). Briefly, partial medium changes were performed daily, and adherent cells were removed by daily transfer into fresh culture dishes. Outgrowth of erythroid progenitor cells (EPCs) was monitored by cytospin preparations and stainings according to May–Gruenwald–Giemsa (Biomed Labordiagnostik GmbH, Oberschleißheim, Germany) and by analyzing the CASY cell counter profiles at days 4, 8, 12, and 14. As soon as erythroid cells start to dominate the culture (days 6–10), the cell concentration was adjusted to 2 × 106 cells/ml and maintained at this value by daily partial medium replacements. At day 14, more than 95% of the cells show the characteristics of EPCs in hematological staining, cell diameter, and flow cytometry analysis (>95% of the cells are positive for CD71 and CD36). Flow cytometry for surface marker analysis was performed with live cells freshly harvested from the cultures and washed once with PBS. Approximately 2 × 105 cells were stained per approach. Antibodies were diluted according to the manufacturer's instructions. Stainings were performed in approximately 50 μl of buffer (PBS supplemented with 0.2% BSA and 1 mM EDTA) for 30 min at 4°C. After staining, the cells were washed once with 1 ml of buffer and resuspended in approximately 400 μl of buffer for measurement. The corresponding isotype controls were used to control for unspecific antibody binding. A minimum of 10,000 events were collected for every staining. The following antibodies were used: anti-CD3 (clone UCHT1), anti-CD34 (clone 581), anti-CD15 (clone 80H5), anti-CD19 (clone J3-119), and anti-CD56 (clone N901) from Beckman-Coulter; anti-CD14 (clone MϕP9), anti-CD36 (clone CB38), anti-CD45 (clone HI30), anti-CD71 (clone M-A712), anti-CD117 (clone YB5.B8), anti-CD235A (clone GA-R2/HIR2), and anti-CD42b (clone HIP1) from BD Biosciences.
Colony-Forming Assays (CFU)
For CFU assays, approximately 100,000 MNCs were diluted to 500 μl with Iscove's modified Dulbecco's medium (IMDM) supplemented with 2% bovine serum albumin (BSA; both from STEMCELL Technologies, Vancouver, Canada). Four hundred microliters of this dilution was mixed with 3.6 ml of methylcellulose medium supporting erythroid burst-forming unit (BFU-E), erythroid colony-forming unit (CFU-E), granulocyte macrophage colony-forming unit (CFU-GM), and granulocyte, erythrocyte, monocyte/macrophage, megakaryocyte (CFU-GEMM) colony growth (STEMCELL Technologies). With a 5-ml syringe and a 16-gauge needle (both BD Biosciences), aliquots of 1.1 ml methylcellulose were seeded in triplicates into 35-mm cell culture dishes (Nunc, Roskilde, Denmark). Approximately 20,000 MNCs were seeded per dish. Cells in methylcellulose were cultivated for 14 days, and colonies were then scored using an inverted microscope (Olympus, Tokyo, Japan) and 4 × and 10 × objectives.
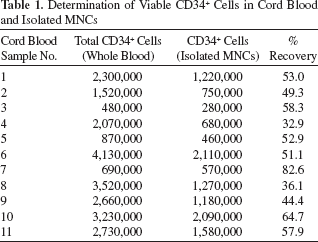
Determination of Viable CD34+ Cells From Fresh Cord Blood Units and Mononuclear Cells
For quantification of viable CD34+ cells from cord blood, an aliquot of 50 μl was mixed with 20 μl of CD45/CD34 staining solution (BD Biosciences, Franklin Lakes, NJ, USA) in TruCount tubes (BD Biosciences) according to the manufacturer's instructions. Cells were stained for 15 min at RT in the dark, and red blood cells (RBCs) were then lysed by addition of 1 ml of ammonium chloride lysing buffer (BD Biosciences). After RBC lysis, 20 μl of 7-aminoactinomycin D (7-AAD; BD Biosciences) dye was added to exclude dead cells. Flow cytometry analysis was performed on a FACScan, and the viable CD34+ cells were quantified using a modified International Society of Hematotherapy and Graft Engineering (ISHAGE) protocol (1, 26). Gating for the different cell types (granulocytes, lymphocytes, monocytes) was performed. The staining protocol was calibrated with the BD Stem Cell Control Kit (BD Biosciences).
For determination of CD34+ cells from isolated MNCs, the cells were diluted with 1 ml of PBS with 0.1% BSA after staining (RBC lysis not necessary), mixed with 7-AAD, and analyzed as described above.
Statistical Analysis
The paired Student's t test was performed to assess the significance of differences between the measurements at different time points. Correlations between independent variables were analyzed using Pearson's product moment correlation. The analysis was performed with SigmaPlot 12 (Systat Software, San Jose, CA, USA).
Results
To examine whether xeno-free cryopreservation of cord blood stem/progenitor cells is an alternative to current protocols using either human or bovine serum, we decided to thoroughly evaluate the performance of the serum-free cryopreservation protocol on 11 cord blood units by four different tests (Fig. 1). First, we directly measured the clinically relevant numbers of viable CD34+ cells before and after cryopreservation. Second, we analyzed total MNC numbers and cell viability before and after cryopreservation. Third, we tested the multilineage differentiation properties of the cord blood-derived cells by performing CFU assays before and after cryopreservation. Fourth, we performed well-characterized erythroid progenitor cell (EPC) expansion cultures before and after cryopreservation. Eleven cord blood units were collected, and an aliquot of 50 μl of each cord blood was removed for determination of cells expressing CD34. MNCs were isolated and resuspended in IBMT medium as described in Materials and Methods. Cells were frozen and stored for either 1 week or 1 year. Reference cells were directly assayed for CD34 expression and total cell viability, and CFU assays and expansion cultures of EPCs were started as shown in Figure 1. The corresponding experiments were performed with the cryopreserved cells after 1 week and 1 year of storage, respectively. Viable CD34+ cells in whole cord blood were quantified using a single-platform method and a modified ISHAGE gating strategy (1) and the numbers of cells expressing CD34 in whole blood and after isolation of MNCs were determined (Table 1). As expected, there is a high variation in the numbers of cells expressing CD34 in the different cord blood samples. For morphological analysis, the isolated MNCs from the 11 CB samples were quantified using a CASY cell counter, and cytocentrifuged onto glass slides for hematological staining (Fig. 2A). The isolated MNCs varied substantially for the different cord blood units, with diverse proportions of lymphocytes, monocytes, granulocytes, and precursor cells as shown in Figure 2A. These differences between the different CB samples were additionally analyzed by flow cytometry, using CD45 staining and SSC (side scatter) properties to gate lymphocyte, monocyte, and granulocyte populations (Fig. 2B). In most samples, granulocytes were strongly depleted when comparing MNCs to whole cord blood. On the other hand, a subset of samples still contained substantial amounts of granulocytes after density centrifugation and red blood cell lysis. These cells were also present after cryopreservation and most probably represent immature granulocyte progenitors, which do not have a higher cell density and cannot be separated from the bulk lymphocytes by Ficoll centrifugation. Immature granulocytes (often characterized by hyposegmented nuclei) can also be identified in the hematological stainings (see arrowheads in Fig. 2A). Figure 3A shows box-whisker plots comparing the recovery of CD34+ cells after cryopreservation relative to the nonfrozen reference cells, which were set to 100%. The recoveries of viable CD34+ cells after 1 week or 1 year of cryopreservation were, with the exception of sample 2 (after 1 week), higher than 75%. Mean recovery for the 11 samples was 90% and 98% after 1 week and 1 year, respectively, with standard deviations of 18% and 16% (see Table 2). The differences in CD34+ numbers after cryopreservation for 1 week or 1 year to the noncryopreserved samples were not statistically significant (p > 0.05, paired t test). For the recovery of total MNCs, again, no significant difference was observed between the 1 week and the prefreeze time point (Fig. 3B) (p > 0.05, paired t test). The recoveries between 1 week and 1 year appear to be different. A closer inspection of the values revealed that this difference is caused by the variation in the cell counts that are used for calculating the cell recovery [the product of cell number (measured with the CASY counter) multiplied by the percentage of viable MNCs (determined by flow cytometry)]. Taking only the cell viability into account, this difference cannot be confirmed (data not shown). Thus, the viability of the cells is not influenced by the storage time, but more accurate cell count measurements are necessary for future analysis.
Schematic representation of the experimental setup and of the analyses performed with the 11 cord blood (CB) samples. After isolation of mononuclear cells (MNCs), aliquots of the cells were directly assayed for total cell viability and colony-forming unit (CFU) capability in triplicates. Viable CD34+ cells were quantified and endothelial progenitor cell (EPC) cultures were initiated. The other aliquots were frozen as described in Materials and Methods. After 1 week or 1 year of storage, cells were thawed and the indicated tests were performed with the cryopreserved samples. Heterogeneity of MNC preparations from different CB units. (A) Aliquots of the isolated MNCs were cytocentrifuged onto glass slides and stained as described (5) in Material and Methods. Pictures were taken at 200 × magnification. Varying amounts of immature progenitor cells are detected besides lymphocytes in the different samples. Immature granulocyte precursors in samples 1, 3, and 8 are marked with arrowheads. Scale bar: 100 μm. (B) Flow cytometric analysis of two representative MNC samples, one with a high (sample 1) and one with a low proportion of immature granulocytes (sample 6). Dot plots for CD45-fluorescein isothiocyanate (FITC) versus side scatter (SSC) are shown. The different leukocyte populations were gated as outlined in (B). Gate 2: lymphocytes, Gate 3: monocytes, Gate 4: granulocytes, Gate 5: CD45dim, SSC low. Analysis of viable CD34+ cells and viable MNCs after cryopreservation. Detailed values and standard deviations are given in Table 2. (A) Box-whisker plot for the recovery of viable CD34+ cells after cryopreservation. The cell numbers prefreeze were set to 100%. Median (thin black line) and mean values (thick gray line) are given in the box. No statistically significant differences were detected between viable CD34+ numbers in the samples before freezing and after 1 week or 1 year of cryopreservation when comparing the mean values (paired t test, p > 0.05 for all comparisons). (B) Box-whisker plot for the recovery of total MNCs after cryopreservation. Total cell number of prefreeze MNCs was set to 100%. Median values for recovery of total MNCs were close to 90% after 1 week or 1 year of cryopreservation. No statistically significant differences were detected between viable MNCs before freezing and after 1 week of cryopreservation when comparing the mean values (paired t test, p > 0.05). Outliers were not removed for the reason of completeness. Statistical tests were also performed after removal of outliers with equal results. Determination of Viable CD34+ Cells in Cord Blood and Isolated MNCs Recovery of Viable CD34+ Cells and MNCs After Short- and Long-Term Cryopreservation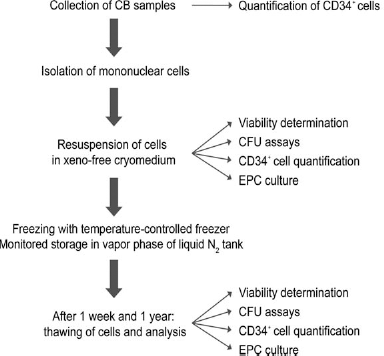



With the exception of CB sample 7, recovery of MNCs was always higher than 80% (Fig. 3B). Table 2 summarizes the results for the recovery of viable CD34+ cells and MNCs after cryopreservation depicted in Figure 4A and B. Taken together, the results show that the xeno-free cryomedium can provide an effective protection of cord blood cells against cryodamage in short- and long-term storage.
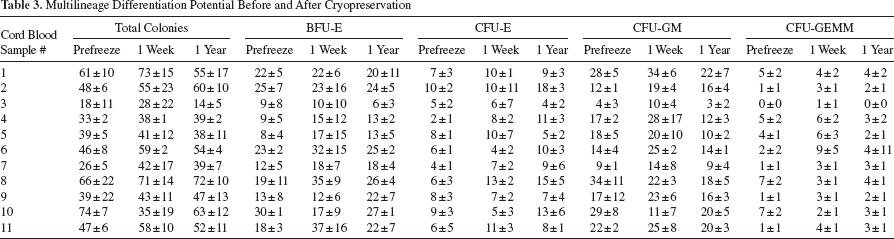
In addition to the determination of cell viability, we also determined the multilineage differentiation potential before and after cryopreservation in IBMT medium by performing colony-forming unit (CFU) assays. We used a semisolid methylcellulose medium, which supports the growth of CFU-E, BFU-E, CFU-GM, and CFU-GEMM colonies. For the majority of the samples, we did not observe statistically significant differences between total colony numbers prefreeze and after 1 week or 1 year of cryopreservation (Table 3). Also, when analyzing the different colony types separately, differences between prefreeze, 1 week frozen, or 1 year frozen samples were not statistically significant due to the inherent variability of the assay, which was performed in triplicates for each time point. Therefore, we can conclude that the multilineage differentiation potential of the cord blood-derived stem/progenitor cells was highly conserved after xeno-free cryopreservation of 1-week or 1-year duration.
In addition, we also analyzed whether short- and long-term cryopreservation affected the expansion cultures of EPCs. Therefore, we expanded EPCs from nonfrozen and cryopreserved samples in serum-free medium and under standardized conditions for 14 days. Expansion of EPCs was successful for all 11 cord blood samples at all tested time points (prefreeze, 1 week, and 1 year cryopreservation), with culture yields from 13 million to 161 million cells starting from 1 × 107 MNCs each (see Table 4). Comparison of the CFU assay data with the relative EPC culture yields, by depicting the mean relative contributions of the four different colony types (CFU-E, BFU-E, CFU-GM, and CFU-GEMM) to the total colony numbers, demonstrates that the EPC culture yield does not strictly correlate with BFU-E and CFU-E numbers, which often were increased after cryopreservation (Fig. 4). In nine cases, there was a reduction in culture yield for the cryopreserved cells. For samples 6 and 8, conversely, we observed an increase of culture yield after 1 week of cryopreservation (samples 6 and 8) or 1 year of cryopreservation (sample 8). Interestingly, the samples with the highest proportion of granulocytes (SSC high gate, see Fig. 2) showed the lowest culture yields in the EPC expansion cultures, irrespective of the numbers of CD34+ cells (Table 4, samples 1, 3, and 8). To demonstrate that the properties of the expanded cells are not affected by cryopreservation, we analyzed the expanded cells at day 14 by hematological staining and flow cytometry for expression of the characteristic markers CD71, CD36, glycophorin A (CD235a), and c-Kit (CD117). EPCs recovered at day 14 are morphologically equivalent and indistinguishable for prefreeze samples and samples cryopreserved for 1 week or 1 year (Fig. 5A). This could also be confirmed by flow cytometry analysis (Fig. 5B). All EPC cultures displayed strong staining for CD71 and CD36, independent of cryopreservation. Variable staining, spanning three orders of magnitude, was observed for glycophorin A (CD235a), indicating that the EPC cultures contain different maturation stages. Again, no differences were observed for the corresponding prefreeze samples and the samples cryopreserved for 1 week or 1 year. All cultures analyzed displayed weak staining for the c-Kit receptor CD117, which is another characteristic feature of EPCs (5). To test if the EPC cultures contain nonerythroid cells, we performed staining with a panel of markers for the other blood lineages (Fig. 5C). As expected, all cells express the common leukocyte antigen CD45. Expression of the hematopoietic stem cell marker CD34, the monocyte/macrophage marker CD14, the B-cell marker CD19, the T-cell marker CD3, the granulocyte marker CD15, the platelet/megakaryocyte marker CD42b, and the natural killer (NK) cell marker CD56 was not observed, underscoring the purity of the expanded EPCs.

Detailed analysis of the results for the CFU assays. Relative contributions of erythroid colony-forming unit (CFU-E), erythroid burst-forming unit (BFU-E), granulocyte macrophage colony-forming unit (CFU-GM), and granulocyte, erythrocyte, monocyte/macrophage, megakaryocyte colony-forming unit (CFU-GEMM) (pie diagrams) and relationship to relative EPC culture yield for each CB sample (step diagrams). EPC culture yields are given in Table 4. Values of prefreeze samples (p) were set to 100%; relative changes after 1 week (w) or 1 year (y) cryopreservation are shown as step diagrams. A relative decrease in CFU-GM and a concomitant increase in BFU-E and CFU-E were frequently observed, indicating that granulocyte and macrophage precursors are more susceptible to damage during cryopreservation.
Multilineage Differentiation Potential Before and After Cryopreservation
Culture Yield Prefreeze/1 Week/1 Year Cryopreservation

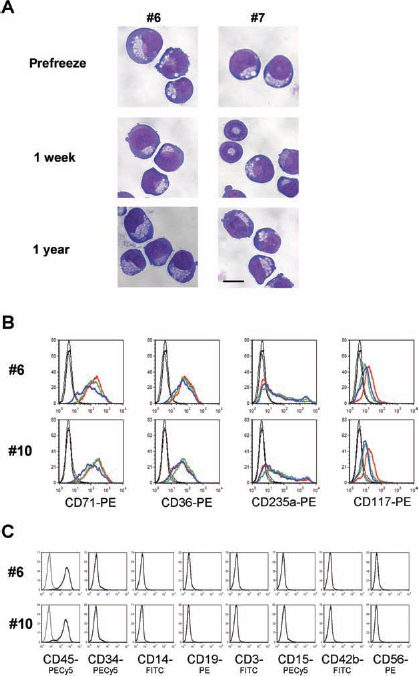
Comparison of EPCs at day 14 of expansion from prefreeze samples and cryopreserved samples. (A) Hematological stainings of cytocentrifuged EPCs from two representative CB units. No difference was observed in cell morphology. Pictures were taken with 600 × magnification. Scale bar: 10 μm. (B) Flow cytomerty analysis of surface marker expression (CD71, CD36, CD235a, and CD117) on EPCs at day 14 from two representative CB units. Thin black lines, isotype controls; red lines, prefreeze samples; green lines, 1 week cryopreserved; blue lines, 1 year cryopreserved. (C) Analysis of the expression of multiple nonerythroid lineage markers on EPC cultures. Markers used were CD45 (pan leukocytes), CD34 (hematopoietic stem/progenitor cells), CD14 (monocytes/macrophages), CD19 (B lymphocytes), CD3 (T lymphocytes), CD15 (granulocytes), CD42b (platelets/megakaryocytes), and CD56 [natural killer (NK) cells]. Antibodies were conjugated to different fluorophores as indicated. Corresponding isotype controls were used for all antibody staining. Thin black lines, isotype controls; thick black lines, marker antibodies.
Discussion
Most recently, we have identified a fully defined, xeno-free cryopreservation medium (IBMT medium) that yielded excellent recoveries after cryopreservation of different stem/progenitor cell types (29). We analyzed whether this xeno-free medium can also be used for efficient cryopreservation of therapeutically relevant cord blood cells and whether the long-term cryopreservation causes cell damage. Here we isolated MNCs from 11 different CB units and compared essential parameters such as CD34+ cell recovery, CFU numbers, and total MNC recovery as well as a well-defined EPC expansion culture (5, 14) before cryopreservation and after 1 week and 1 year of storage in liquid N2 (Fig. 1). Interestingly, a big proportion of CD34+ cells is already lost during Ficoll gradient centrifugation and subsequent erythrocyte lysis during the isolation of MNCs (Table 1). This loss of CD34+ cells was not critical as it only reduced the starting basis for the subsequent analysis of the effect of xeno-free cryopreservation. We found excellent recoveries of CD34+ cells and total MNCs (Fig. 3). The overall capability to form CFUs of the cryopreserved samples was as good or even better as the one of the prefreeze samples, while the composition of CFUs slightly varied (Table 3 and Fig. 4). Thus, our data indicate that this fully defined medium is an excellent alternative to serum-containing formulations and that the multilineage differentiation potential of the cells is not compromised. Recently, Page et al. reported that total CFU numbers are a strong predictor of neutrophil and platelet engraftment (20). This single center analysis of 435 cord blood transplants showed that CFU numbers after thawing were more predictive for successful engraftment than CD34+ cell numbers or total nucleated cells. The study was performed in a single transplantation center with CB units from different CB banks. Interestingly, in this large clinical setup, recovery of CFUs after cryopreservation was very low compared to our results (median recovery 21.2%). In our setup, we did not observe a statistically significant difference between the CFU assays performed with fresh or cryopreserved cells (see Table 3). CFU assays are difficult to standardize, as they involve manual counting of morphologically identified colonies. Wagner et al. also reported big discrepancies between the CFU values reported by different cord blood banks and the values determined in the transplantation center (28). For our study, all CFU assays were set up and scored by one person, and the same lot of methylcellulose medium (with a shelf-life which extended beyond the 1-year storage period) was used for all assays to minimize variation. In addition, our study was performed with a Ficoll-purified MNC fraction, and residual erythrocytes were removed by a mild lysis step, resulting in a very pure cell suspension. Therefore, our samples and the general setup, located in a basic research laboratory, strongly differed from the situation in the study of Page et al. Nevertheless, it is an exciting possibility that the high recovery of CFUs in our samples is due to the IBMT medium. Of course, further tests on larger sample numbers are required with the IBMT cryomedium to assess its safety and feasibility for clinical use. We also performed successful EPC expansion cultures for all cord blood samples from aliquots of 1 × 107 MNCs (Fig. 5 and Table 4). Culture yield declined after cryopreservation of the samples with two exceptions, for which we observed an increase after cryopreservation. Neither numbers of recovered viable CD34+ cells nor BFU-E numbers correlated well with the EPC culture yield before cryopreservation or after cryopreservation. Interestingly, van den Akker et al. recently reported that the majority of erythroid expansion potential resides in the CD34-negative fraction (27) under very similar culture conditions, which could explain the missing correlation of culture yield to CD34+ numbers. The BFU-E progenitors are also considered to still express the CD34 antigen (24, 25). Thus, the identity of the progenitor cells, which give rise to the majority of EPCs, is not fully understood up to now. Based on our results, we speculate that these progenitors are more sensitive to the cryopreservation process than the CD34+ cells. We cannot exclude that the frequent manipulation steps of the cultures (like the daily feeding and replating) introduced a bias to our analysis. Therefore, we consider the successful EPC cultures as a qualitative measure of progenitor cell expansion potential. Our data imply that high numbers of immature granulocytes, which are only inefficiently removed during Ficoll centrifugation, negatively affect the EPC outgrowth (Figs. 2, 4 and Table 4). Therefore, a further purification step, like isolation of CD34+ cells or CD133+ cells from cord blood, could be useful to avoid these inherent heterogeneities for quantitative experiments. Detailed statistical analysis revealed only a weak correlation between the culture yield with freshly isolated cells and cells after 1 week of cryopreservation (p < 0.05, correlation coefficient 0.609), whereas culture yields with 1 week and 1 year cryopreserved samples were highly correlated (p < 0.001, correlation coefficient 0.853). This indicates that freezing of the cells clearly has a strong impact on the expansion cultures, whereas the time of storage has less impact.
Recent reports indicate that hematopoietic stem/progenitor cells are well conserved after long-term storage of cord blood units (3, 4). These encouraging results underscore the importance of cord blood as an alternative stem cell source. Therapy success of CBT infusion was shown to clearly correlate with the number of infused CD34+ cells (22), underscoring that optimization of the cryopreservation procedure is clearly of major importance for future therapeutic use of cord blood cells. The feasibility of transfusion of highly purified HSCs has also been demonstrated in clinical trials with cancer patients in autologous settings (17, 18), but this approach is not widely accepted so far, due to the challenge of purifying HSCs with a cell sorter under clinical, good manufacturing practice (GMP)-compliant conditions. The benefits of purified HSC grafts compared to conventional mobilised peripheral blood mononuclear cell (PBMC) grafts are also not clear yet, as the time to reach neutrophil and platelet recovery could be prolonged due to the absence of differentiated progenitor cells (17). These drawbacks could be overcome by using in vitro expanded stem/progenitor cell grafts, which could provide long-term engraftment through HSCs and short-term immune recovery through progenitor cells, which are inevitably produced during HSC expansion. Since the storage of cord blood samples for 1 year did not show any cryodamage, we are confident that this xeno-free medium is a true alternative to serum containing formulations. Additionally, we also expect that in vitro expanded stem cells can be efficiently cryopreserved for extended time periods.
Moreover, regulatory rules and guidelines for the quality control of frozen cell samples for medical applications, as stated by the EC-directive 2004/23/EG and by the FDA in the United States, also require optimization and standardization of cryopreservation protocols and storage conditions and avoidance of xeno-materials for therapeutic applications.
In summary, we could demonstrate that a fully xeno-free cryopreservation protocol can be applied to cord blood-derived cells with excellent recovery of viable CD34+ cells and total MNCs after short- and long-term cryopreservation. In addition, the multilineage differentiation potential of the stem/progenitor cells, as analyzed by CFU assays, and the capability to perform mass expansion cultures were not compromised by long- and short-term cryopreservation. Thus, our xeno-free procedure provides a novel highly demanded tool for the cryopreservation and subsequent use of cord blood as a source for stem cells.
Footnotes
Acknowledgments
This work was supported by grants from the European Commission (FP6-project: LSHB-CT-2006-037261 “CRYSTAL,” Cryopreservation of stem cells for human therapeutic application) to H. Zimmermann and A. Kolbus. We thank Marina Schorpp-Kistner for critically reading the manuscript. J. C. Schulz and H. Zimmermann declare the following conflict of interest: After the present study, the composition and recipe of IBMT medium have been acquired from Rene Fischer (Fischer Procryotect, Ruedlingen, Switzerland) and licensed.