
Abstract
Cancer stem cells (CSCs) in glioblastoma multiforme (GBM) are radioresistant and chemoresistant, which eventually results in tumor recurrence. Targeting CSCs for treatment is the most crucial issue. There are five methods for targeting the CSCs of GBM. One is to develop a new chemotherapeutic agent specific to CSCs. A second is to use a radiosensitizer to enhance the radiotherapy effect on CSCs. A third is to use immune cells to attack the CSCs. In a fourth method, an agent is used to promote CSCs to differentiate into normal cells. Finally, ongoing gene therapy may be helpful. New therapeutic agents for targeting a signal pathway, such as epidermal growth factor (EGF) and vascular epidermal growth factor (VEGF) or protein kinase inhibitors, have been used for GBM but for CSCs the effects still require further evaluation. Nonsteroidal anti-inflammatory drugs (NSAIDs) such as cyclooxygenase-2 (Cox-2) inhibitors have proven to be effective for increasing radiation sensitivity of CSCs in culture. Autologous dendritic cells (DCs) are one of the promising immunotherapeutic agents in clinical trials and may provide another innovative method for eradication of CSCs. Bone-morphogenetic protein 4 (BMP4) is an agent used to induce CSCs to differentiate into normal glial cells. Research on gene therapy by viral vector is also being carried out in clinical trials. Targeting CSCs by eliminating the GBM tumor may provide an innovative way to reduce tumor recurrence by providing a synergistic effect with conventional treatment. The combination of conventional surgery, chemotherapy, and radiotherapy with stem cell-orientated therapy may provide a new promising treatment for reducing GBM recurrence and improving the survival rate.
Keywords
Introduction
Glioblastoma multiforme (GBM) is the most common type of primary malignant brain tumor, accounting for 55% of primary brain tumors (43). The prognosis of GBM is very poor; most patients die of tumor recurrence (39,70). The best mean survival time with successful tumor resection, radiotherapy, and temozolamide (Temodal, TMZ) chemotherapy may reach up to 18 months (17,51,70). But even in the best of the most recent reports, the 2-year survival rate was still less than 30% (67,68). Causes of recurrence are complex and include no clear tumor margin for complete resection, high proliferative index, resistance to chemotherapy and radiotherapy especially in cancer stem cells (CSCs), and cerebrospinal fluid (CSF) dissemination.
Recently, targeting cancer stem cells (CSCs) has emerged as a treatment (1–5,7,12,16,23,25,35,60). The CSCs have a multipotent function, having self-renewal potential and resistance to chemotherapy and radiotherapy (6). Liu et al. (45) reported that the resistance was probably contributed to by CD133+ cells with higher expression of breakpoint cluster region pseudogene 1 (BCRP1; drug resistant gene) and O-6-methylguanine-DNA methyltransferase (MGMT; DNA repair enzyme) as well as antiapoptosis proteins and inhibitors (37). In comparison to CD133-cells, CD133+ cells have 32- to 56-fold more activity of MGMT. Ji et al. (36) showed that CD133+ CSCs express high levels of the drug transporter gene BCRP, combined with upregulation of DNA repair protein, MGMT mRNA, and mRNAs of other genes that inhibit apoptosis including FAS-associating death domain (FADD)-like antiapoptotic molecule (FLIP), B-cell CLL/lymphoma 2 (Bcl-2), Bcl-X, and some inhibitor of apoptosis (IAP) family members (55,57,61). Nakai et al. (53) showed CSCs possess stronger drug resistance to conventional anticancer drugs such as doxorubicin, etoposide (VP-16), carboplatin, and bis-chloroethylnitrosourea (BCNU) than non-stem cells of malignant gliomas. Double immunofluorescence staining showed coexpression of multidrug resistance 1 (MDR1) and CD133 on CSCs. Johannessen et al. (37) reported that the alkalizing agents BCNU and TMZ are the drugs of choice for adjuvant glioma chemotherapy. A DNA repair mechanism can restore the integrity of alkylated DNA bases and thus contribute to drug resistance. Cancer stem cells possess enhanced DNA repair capacity (37).
Recently, there are many methods for influencing the growth of CSCs. TMZ is effective for treating patients with positive methylated promoter of MGMT based on clinical experience. Fu et al. (24) reported CSCs display a strong capability of tumor resistance to TMZ. This resistance was probably contributed to by CD133+ cells with down-regulation of autophagy-related proteins. Beier et al. (8) reported TMZ induced a dose- and time-dependent decline of brain CSCs in a cell culture study. TMZ showed some effectiveness in reduction of CSCs at the level of 50 μmol/L and was most effective at the level of 500 μmol/L. However, with the usual clinical dose, the CSF level of TMZ only reaches 50 μmol/L.
In conventional radiation, undamaged CD133+ cells exhibit activating phosphorylation of three key mediators of cell cycle checkpoints—Rad17, Chk1, Chk2—that are significantly enhanced after exposure to radiation (53). In CD133- cells, these proteins are phosphorylated only in response to ionizing radiation, and the magnitude is blunted compared with CD133+ cells. This implies that glioma stem cells exhibit constitutive activation of multiple cell cycle checkpoints that may further upregulate in response to DNA damage. Chalmers (12) showed that the radioresistant phenotype might be the consequence of constitutively upregulated and hyperresponsive cell checkpoint pathways.
Usually, CSCs constitute only 2–3% of a glioma tumor (23). The most commonly used tumor marker is CD133+ (a membrane protein of CSCs and other cells). Singh et al. (65) reported that even 100 CD133+ cells transplanted into severe combined immunodeficient (SCID) mouse brain can reproduce a new GBM tumor mass but 106 cells of CD133- cannot create the same GBM tumor mass. On the other hand, the CSC count is correlated with tumor malignancy (69). The mean CSC count for malignant astrocytoma (AA) is somewhat less than 1%, but for GBM it may increase to 3–5%, as shown in Figure 1. Usually the CSCs are in a quiescent state. But when they are stimulated by surgery, chemotherapy, and radiotherapy, their population may grow exponentially (46). When CSCs regrow rapidly, patients eventually die of tumor recurrence. Targeting the CSCs for treatment is an innovative therapeutic rationale (42). Targeting this small portion of tumor consisting of cancer stem cells may improve patient survival.

The percentage of cancer stem cells (CD133+) in a glioblastoma multiforme (GBM) tumor is significantly higher than in an anaplastic astrocytoma (AA) tumor.
In ongoing multimodal and pluridisciplinary treatment of GBM with surgery and subsequently radiation and chemotherapy (32,42), most tumor cells are eradicated but the residual CSCs usually increase as shown in Figure 2. Furthermore, the recurrence is unavoidable. Hermann et al. (32) showed that a novel approach targeting CSCs is capable of overcoming resistance to therapies. Therefore, initially reducing CSCs should be most crucial.

Cancer stem cells may be transformed from normal stem cells or progenitor cells or from glial cells. Radiation and chemotherapy may eradiate most of tumor cells and increase the population of radioresistant and chemoresistant cancer stem cells, respectively.
Currently Targeting CSCs for GBM
Schulenburg et al. (63) reported that neoplastic stem cells might be a novel therapeutic target in clinical oncology. Up to now, there have been five methods used for eradicating CSCs, as shown in Figure 3. One method is focused on targeting a new molecular protein signal pathway of CSCs with a new targeting therapeutic agent. Another method is focused on increasing the radiosensitivity (47) and chemotherapy sensitivity of CSCs by using a hypersensitivity agent (15). A third method involves using immunotherapy including autologous dendritic cells (DCs). A fourth method uses a differentiation agent such as bone morphogenetic protein-4 (BMP-4) to promote the CSCs to differentiate into normal cells. Finally, gene therapy may be used to reduce CSC proliferation.

Five methods currently used to treat CSCs.
Signal pathways targeted in CSCs include wingless-type MMTV integration site family (Wnt) (54), sonic hedgehog (shh) (7), Notch (34), homeobox (HOx) family, B lymphoma Mo-MLV insertion region 1 homolog (Bmi-1) (52), and phosphatase and tensin (PTEN) (19), telomerase (48), efflux transporters (50), and epidermal growth factor (EGF) (20,66), micro-RNA (44) and vascular epidermal growth factor (VEGF) receptors (50). We may be able eliminate CSCs by employing a new targeting therapy. Targeting CSCs by using passive immunotherapy (antibody therapy) such as anti-CD-44 antibody in acute myeloblastic leukemia (AML) or active immunotherapy (72) such as dendritic cells is undergoing investigation in ongoing trials. Also being investigated is the strategy of promoting CSCs to differentiate into normal cells. BMP-4 may induce CSCs to become normal cells (59). Once the CSCs are eliminated, then subsequent conventional chemotherapy and radiation may be more effective.
Targeting CSCs on Oncoprotein Pathway and Receptor
Nicolis (54) reported that partially differentiated precursor cells might develop into neural stem cells through fibroblastic growth factor-2 (FGF-2) and EGF. These neural stem cells may be the origin of cancer. The Shh and Wnt pathways lead to cancer cell proliferation. Fan et al. (22) showed that the Notch signaling pathway regulates normal stem cells in the brain and that GBMs contain stem-like cells with higher Notch activity. Notch blockade by γ-secretase inhibitors reduces neurosphere growth and clonogenicity in vitro. Hovinga et al. (34) also emphasized that the Notch pathway plays a critical role in linking angiogenesis and CSC self-renewal and thus is a potential therapeutic target.
Gal et al. (26) reported that a key hedgehog pathway target was highly expressed in five of 19 primary GBM specimens and in four of seven GBM cell lines. Shh ligand was expressed in GBM-derived neurospheres, suggesting a mechanism for pathway activation. Bar et al. (7) reported that cyclopamine might block the hedgehog pathway causing a 40–60% reduction in growth of glioma cell lines.
Gilbertson et al. (29) reported that CSCs can secrete diffusible factors such as VEGF, which recruits aberrant tumor vasculature to the niche, making these cells a therapeutic target. Clinical trials of the antiangiogenic drugs bevacizumab and cediranib have demonstrated encouraging efficacy in patients with GBM. This antitumor effect could be the result of normalization of tumor vasculature or depletion of tumor blood supply. These drugs might function by disrupting stem cell maintenance. Calabrese et al. (10) showed that treating glioblastoma-bearing mice with bevacizumab depleted tumor blood vessels and caused a dramatic reduction in the number of GBM stem cells and the growth rate of tumors.
Borovski et al. (9) reported that tumor microvasculature supported proliferation and expansion of glioma-propagating cells. CD133+ CSCs are located in the perivascular niche and interaction with the niche promotes tumor growth and therapy resistance. Microvascular endothelial cells support the growth of glioma propagating cells (GCPs) resulting in a higher tumor proliferation rate. Aguado et al. (1) reported that cannabinoids not only induce apoptosis of transformed cells but inhibition of tumor angiogenesis also induces glioma stem cell differentiation and inhibits glioma angiogenesis.
The efficacy of targeted therapies that inhibit tyrosine kinase pathways in GBM has been modest. CD133+ cells express higher levels of BCRP1 and MGMT mRNA as well as higher mRNA levels of genes that inhibit apoptosis. CD133 cancer cells promote tumor angiogenesis through increased expression of VEGF. However, Gadji et al. (25) showed that monotherapy with a tyrosine kinase inhibitor (EGF receptor-targeted inhibition) has no therapeutic effect. Bao et al. (6) showed that a multidirectional drug approach for CSCs plays a role in promoting angiogenesis and that this action is blocked with the anti-VEGF antibody bevacizumab.
Targeting the CSCs by Radiation and Chemotherapy Sensitizers
Sauvageot et al. (62) reported that heat shock protein (HSP-90) has the ability to inhibit CSCs and synergize with radiation and or TMZ. Michelakis et al. (50) also reported that anti-epidermal growth factor receptors (cetuximab, nimotuzumab) increase the radiosensitivity of CSCs. Stea et al. (66) found that the time- and dose-dependent radiosensitization of GBM exists because of the EGF receptor tyrosine kinase inhibitor (Iressa). Kang et al. (38) reported that, when combined with BCNU, a chloride channel blocker might promote apoptosis or sensitize gliomas to BCNU chemotherapy. Xu et al. (72) also found that CD44 is a major cell surface hyaluronan receptor and cancer stem cell marker. CD44 is upregulated in GBM, and its depletion blocks GBM growth and sensitizes GBM cells to cytotoxic drugs in vivo. CD44 antagonists potently inhibit glioma growth in preclinical mouse models.
Martin et al. (49) demonstrated advances through antiangiogenesis therapy for gliomas. Targeting the inflammatory cascade with a cyclooxygenase-2 (Cox-2) inhibitor that plays a role in glioma angiogenesis, decreased tumor cell proliferation and tumor size, and increased tumor apoptosis. Celecoxib and other nonsteroidal antiinflammatory drugs (NSAIDs) (11,14) significantly enhanced GBM radiosensitivity and prolonged the survival of GBM implant mice by inhibiting tumor angiogenesis, resulting in extensive tumor necrosis.
Targeting CSCs by Immunotherapy
Ghebeh et al. (28) reported that cancer stem cell immunotherapy is the right bullet for the target. Immune suppression is commonly found in patients with GBM including decreased cellular and humoral immunity. Pellegatta et al. (56) reported that neurospheres enriched in CSCs were highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas.
Cancer stem cells express high levels of tumor-associated antigens as well as major histocompatibility complex molecules. Xu et al. (71) reported that DC vaccination using CSC antigens elicited antigen-specific T-cell responses against CSCs. Dendritic cell vaccination-induced interferon-γ production is positively correlated with the number of antigen-specific T-cells generated. In our laboratory, we found that cytotoxic T lymphocytes may kill the CSCs in cell culture (Fig. 4). The survival rate of neurosphere lysed-pulsed dendritic cells was longer than in that of the control group.
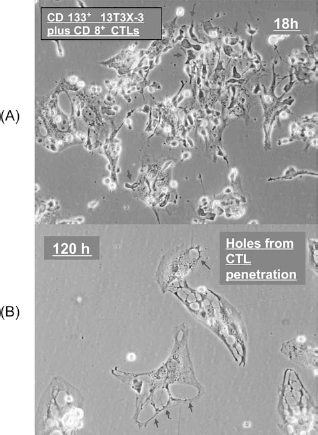
(A) Microscopic field shows cytotoxic T lymphocytes (CD8+) (arrows) cocultured with CD133+ cancer stem cells at 18 h. (B) After 120 h of incubation, CD8+ T lymphocytes attacked the CD133+ cancer stem cells. The CD133+ cancer stem cells were destroyed by lymphocytes. The cell membranes of the cancer stem cells were ruptured, and their cytoplasm was vacuolated (arrows).
Two vaccine preparations have been tested, mixed leukocyte culture (MLS) and tumor B-cell hybridoma (THB). Yao et al. (73) found that B7 homolog 1 (B7-H1 or programmed cell death ligand) was positively correlated with malignancy grade gliomas and negatively correlated with tumor infiltration CD8+ T-cells but was not expressed exclusively on tumor stem cells. B7-H1 is the third member of the B7 family that has been described to negatively regulate T-cell functions.
Hongeng et al. (33) reported that CD3+ CD56+ cytokine-induced killer (natural killer, NK) cells appear to be a promising effector cell type but with great cytotoxicity for pediatric cancer cells.
Ahmed et al. (2) showed that stimulation of human epidermal growth factor receptor 2 (HER2)-specific T-cells with HER2-positive autologous GBM cells results in T-cell proliferation and secretion of INF-γ and interleukin-2. HER2-specific T-cells kill CD133+ and CD133- cells derived from primary HER2-specific GBM. Duhem-Tonnelle et al. (21) showed that erb3 (HER2-new) immunoreactivity was prominent in CD133+ putative tumor stem cells, which suggests that erb3 may represent a new potential therapeutic target.
Targeting CSCs by Promoting Cell Differentiation
Altaner (4) showed that CSCs contribute to glioma radioresistance by increasing DNA repair capacity through preferential activation of the DNA damage response checkpoints. BMP4 reduces proliferation of CD133+ cells effectively in vitro and tumor growth in vivo (59). BMP4 may act as a key inhibitory regulator of cancer initiation and may be used in combined stem cell-based therapy as a noncytotoxic therapeutic agent. BMP4 treatment also may render the stem cells more susceptible to a conventional therapy. Piccirillo et al. (58) showed that BMP4 can reduce the tumorigenic ability of CSCs in GBM. Mechanisms regulating the physiology of normal brainstem cells may still be in place in cancerous brainstem cells, and this may lead to the development of cures that selectively target the CSCs population.
Targeting CSCs by Gene Therapy
Dey et al. (18) reported on virotherapy, a novel therapeutic approach utilizing conditionally replicative viruses to directly target CSCs (31). In virotherapy, oncolysis is achieved by intraneoplastic cell virus replication-mediated cell lysis. A single-stranded RNA virus is the natural host. Reovirus selectively replicated in glioma cells inhibits RNA-activated protein kinase activation, and thus, viral protein can be synthesized leading to tumor regression. Viral replication and CSC lysis lead to infection of neighboring CSCs (23). Chang et al. (13) reported on using lentiviral vector expressing small hairpin RNA (shRNA) to knockdown sirtuin 1 (SirT1) expression in GBM CD133+ cells. Silencing of SirT1 significantly enhanced the sensitivity of GBM CSCs to radiation and increased the level of radiation-mediated apoptosis, which has been proven in nude mice transplanted with CSCs to cause inhibition of tumor growth and improve the mean survival rate.
Inappropriate met proto-oncogene (c-Met) activation in cancer occurs through autocrine and paracrine activation, transcriptional overexpression, gene amplification, and activating mutations and has been observed in brain tumors. Overexpression of c-Met in brain tumor cells enhances their tumorigenicity, tumor growth, and tumor-associated angiogenesis. Guessous et al. (30) reported that small molecule kinase inhibitor of c-Met may inhibit tumor stem cell malignancy.
Aloy et al. (3) showed that HSP27 has the ability to protect cells from stressful stimuli and increased levels in tumors resistant to anticancer therapeutics. HSP27 gene therapy offers a potential adjuvant therapy to radiation-based therapy of resistant tumors.
Stem cell-based gene therapy using rat mesenchymal stem cells engineered with adenoviral vector encoding human interleukin-2 (IL-2) has been shown to be an effective treatment for glioma cells in in vitro and in vivo experiments. Balyasnikova et al. (5) reported that genetically modified mesenchymal stem cells can express a single-chair antibody against EGFRvIII on the cell surface. Human adult mesenchymal stem cells are cellular vehicles for delivery of antitumor agents to solid tumors providing a new method for enhanced delivery of therapy.
Zhang et al. (74) reported that the transcriptional repressor RE1-silencing transcription factor (REST), which is best known for its role in neural progenitor cells, may also be crucial for self-renewal of CSCs. Gangemi et al. (27) showed that sex-determining region Y box 2 (SOX2) is a master gene involved in sustaining self-renewal of CSCs. Knockdown of SOX2 expression in glioma cells using retroviral vectors harboring a microRNA engineered to target SOX2 mRNA may stop proliferation and result in the loss of tumorigenicity.
Sherry et al. (64) found that small molecule inhibitors of signal transducer and activator of transcription 3 (STAT3) DNA binding inhibits cell proliferation and the formation of new neurospheres from single cells. Genetic knockdown of STAT3 using an shRNA also reduces GBM stem cell proliferation and neurosphere formation.
Gal et al. (26) reported that microRNA (MIR-451) and imatinib inhibit tumor growth of glioblastoma stem cells. Krichevsky et al. (40) found that MIR-21 regulates oncogenic roles and has potential as a disease biomarker and novel therapeutic target in oncology. Marian et al. (48) showed that the telomerase antagonist imetelstat efficiently targets glioma tumor-initiating cells leading to decreased proliferation and tumor growth.
Conclusions
The combination of conventional surgery, chemotherapy, and radiotherapy with stem cell-orientated therapy may provide a new promising treatment for reducing GBM recurrence and improve the survival rate (41) as shown in Figure 5. Targeting stem cells using signal pathway inhibitors, radiosensitizers and chemosensitizers, immunotherapy, stem cell differentiation promoters, and gene therapy may provide a new clinical field.

The combination of cancer stem cell-targeted therapy with conventional therapy may reduce the number of cancer stem cells that remain and reduce tumor recurrence when compared with conventional treatment only.
Footnotes
Acknowledgments
This study was supported in part by the Taiwan Department of Health Cancer Research Center of Excellence (DOH-TD-C-111-005) and in part by the Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH-TD-B-111-004). It also was partially funded by a grant from the National Science Council (NSC-2314-B-039-012-MY2).