
Abstract
Mesenchymal stem cells (MSCs) of bone marrow origin appear to be an attractive candidate for cell-based therapies. However, the major barrier to the effective implementation of MSC-based therapies is the lack of specific homing of exogenously infused cells and overall the inability to drive them to the diseased or damaged tissue. In order to circumvent these limitations, we developed a preconditioning strategy to optimize MSC migration efficiency and potentiate their beneficial effect at the site of injury. Initially, we screened different molecules by using an in vitro injury–migration setting, and subsequently, we evaluated the effectiveness of the different strategies in mice with acute kidney injury (AKI). Our results showed that preconditioning of MSCs with IGF-1 before infusion improved cell migration capacity and restored normal renal function after AKI. The present study demonstrates that promoting migration of MSCs could increase their therapeutic potential and indicates a new therapeutic paradigm for organ repair.
Keywords
Introduction
The last decade has witnessed an explosion of studies exploring the therapeutic potentials of mesenchymal stem cells (MSCs) for a variety of diseases and disorders (3,29). MSCs can be easily isolated from adult tissues and expanded in vitro without losing their phenotypic characteristics, they are free of ethical controversies and most importantly exhibit no significant immunogenicity, which permits allogenic transplantation without immunosuppressive drugs. Although MSCs were initially considered for therapies based on their multilineage differentiation capacity (2,34), the ability to secrete cytokines and growth factors with antiapoptotic and proangiogenic properties (14,26,42,47) and their capacity to reduce scarring and inflammation (46) have positioned MSCs in the focus of a broad range of emerging therapeutics (7,9,13,18,24,33). However, clinical studies targeting various pathologies have repeatedly provided controversial results. For example, the beneficial effects of MSCs to treat myocardial infarction are not consistent when comparing data obtained from short-term (11) and long-term (27) follow-ups, while ongoing clinical trials for graft-versus-host disease (GvHD) presented mixed clinical data (23,28).
Presently, the major limitations that contribute to the failure of clinical trials are mainly the lack of specific homing after systemic infusion, and potentially, the poor survival of MSCs in the proinflammatory microenvironment of injured tissue. Given the low efficacy of MSCs, a high dosing, ranging from 150 million to 300 million cells administered twice per week over the course of 2 weeks, is usually used in clinical trials (17). High dosing entails extensive in vitro expansion, which is known to induce cell enlargement, potentially elevating the risk of entrapment of cells within nonspecific tissues. Thus, a large fraction of systemically infused MSCs becomes massively trapped within the lungs and filtering organs as emboli (4,20). Alternatively, they passively arrest and interrupt blood flow during the first pass through the precapillary level (44), thus preventing the majority of infused MSCs from homing to damaged or diseased tissues.
Various strategies are being developed to reduce the number of administered cells by enhancing cell homing to target tissues or prolonging the presence of the cells in the tissue of interest (45). Genetic modification of MSCs with retroviral vectors encoding homing receptors such as C-X-C chemokine receptor type 4 (CXCR4) or the α4 subunit of the very late antigen-4 (VLA-4)-integrin has been recently used to enhance homing of MSCs (5,8,21). Furthermore, enzymatic conversion of native CD44 glycoform on MSC membrane into hematopoietic cell E-selectin/L-selectin ligand (HCELL) enabled intravenously infused MSCs to home bone marrow more efficiently than unmodified cells (36). Genetically modified MSCs with the serine protease kallikrein by adenovirus transduction were more resistant to oxidative stress-induced apoptosis and enhanced protection against acute ischemic kidney injury by inhibiting apoptosis and inflammation (10). Although genetic modification may be a viable approach to improve cell homing and/or survival, current methods raise potential safety concerns and are technically convoluted and incapable to accommodate a broad range of ligands and factors or are not adaptable to multiple cell types.
Here we attempted to develop an alternative approach to optimize MSC migration efficiency and potentiate their beneficial effect at the sites of the injury. To this direction, we focused on preconditioning MSCs before infusion with various compounds possessing prosurvival and promigratory properties and potentially devoid of side effects. Among the molecules that have been tested for their promigratory properties was Glial cell-derived neurotrophic factor (GDNF), shown to increase the motility and survival of cultured kidney-derived MSCs (39) and tumor necrosis factor-α (TNF-α), reported to improve migration of cultured MSCs towards chemokines (35) and enhance MSC homing into the ischemic myocardium (38). Another possible candidate examined for its promigratory properties was insulin-like growth factor-1 (IGF-1), which proved to mediate the beneficial effect of MSCs on tubular cell repair in acute kidney injury (AKI) (16) and to enhance the migratory capacity of MSCs in vitro (25). Hence, we initially examined the capacity of these molecules to exert promigratory effect on MSCs by using an in vitro injury–migration model, and subsequently, we evaluated the viability of the method in a cisplatin-induced AKI model.
Our results indicated that preconditioning with IGF-1 before infusion markedly increases the number of MSCs homing the injured kidney, thereby ameliorating renal structure impairment and promoting the recovery of renal function.
Materials and Methods
Chemicals were from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise specified.
Isolation and Purification of MSCs
MSCs were obtained from 2-month-old male C57BL6/J mice (Charles River Italia S.P.A., Calco, Italy) as previously described (29). Briefly, mice were killed, and femurs and tibias were aseptically removed. Bone marrow (BM) was flushed from the shaft of the bone with Dulbecco's modified Eagle's medium (DMEM) containing 2% fetal calf serum (FCS; Invitrogen, Paisley, Scotland) plus penicillin/ streptomycin (100 U/ml to 0.1 mg/ml; Invitrogen) and then filtered through a 100-μm sterile filter (BD Falcon, Oxford, UK). Filtered BM cells were plated in DMEM plus 10% FCS and penicillin/streptomycin (100 U/ml to 0.1 mg/ml) and allowed to adhere for 6 h. MSCs were recovered from BM by their tendency to adhere tightly to plastic culture dishes and nonadherent cells were then removed.
After 10–12 days MSCs were immunodepleted of CD45-positive cells as previously described (29). Medium was changed regularly every 3 days; after 10 days, cells were switched to 2% FCS, o/n and incubated with IGF-1 (20 ng/ml; StemCell Technologies, Vancouver, Canada), GDNF (100 ng/ml; Abcam, Cambridge, UK), or TNF-α (100 ng/ml) and detached by trypsin-EDTA (0.5–0.2 g/L; Invitrogen) to be used for the in vivo and in vitro experiments.
Migration Assays
Scrape-Healing Assay
MSCs were seeded at 3.5 × 104 cell/cm2 in medium containing 10% FCS and were allowed to reach confluence. Subsequently, they were switched to 2% FCS and incubated with IGF-1 (20 ng/ml) for 36 h, GDNF (100 ng/ml), or TNF-α (100 ng/ml) for 24 h. The monolayers were then scratched by the tip of a pipette, washed with phosphate-buffered saline (PBS), and incubated in 2% FCS for 20 h in the presence of the factors apart from TNF-α. Images of cell samples were taken by a digital camera. In additional experiments culture medium was removed and adherent cells were harvested and counted 20 h after scratching. For CXCR4 inhibition experiments, MSCs were treated with IGF-1 (20 ng/ml) for 36 h, exposed to CXCR4 neutralizing antibody (5 μg/ml; R&D Systems, Inc., Minneapolis, MN, USA) for 1 h and then submitted to the scrape-healing assay at the presence of the antibody. For individual cell motility analysis, MSCs were cultured in 24-well plates (BD Falcon) and pictures were taken every hour for 20 h using a time-lapse microscope (Carl Zeiss, Jena, Germany) equipped with a humidified chamber with 5% CO2 at 37°C. Analysis of cell motility was performed on sequential time-lapse images with AxioVision software (Carl Zeiss). Briefly, each individual cell was tracked, and the following features were measured: distance (the total distance in pixels traveled from the first to last timepoint), straight distance (the straight line connecting the first and last time point), cellular velocity (the traveled pixels/min).
Transfilter Assay
Cell migration was also studied in Transwell chambers (Corning, Costar Italia) by seeding MSCs (n = 3) unstimulated or pretreated with IGF-1 (20 ng/ml, 36 h) (n = 3), GDNF (100 ng/ml, 24 h) (n = 3), or TNF-α (100 ng/ml, 24 h) (n = 3) on the upper side of a porous polycarbonate membrane (pore size: 8 μm; Euroclone S.p.A., Pero, Italy) in coculture with murine proximal tubular epithelial cells (PTECs). PTECs were provided by Dr. Eric G. Neilson (Vanderbilt University, Nashville, TN) and grown as previously described (16). PTECs were seeded at 4.6 × 104 cells/cm2 and 24 h later incubated with DMEM plus 2% FCS (test medium) alone or in the presence of 2.5 μM cis-platinum (II)-diamine dichloride (cisplatin; Ebewe Italia srl, Rome, Italy) for 6 h. After cisplatin removal, cells were washed and cocultured with MSCs. Twenty-four hours later, the cells at the upper side of the filter were mechanically removed. Cells that had migrated to the lower side of the filter were fixed for 30 min in 11% glutaraldehyde and stained with hematoxylin and eosin (BioOPtica, Milan, Italy). Ten random fields were counted for each filter. The concentration and time of GDNF incubation were chosen on the basis of previous studies (39). Experimental conditions for IGF-1 and TNF-α treatment were based on previous studies (25,35,38) and optimized as follows: MSCs were treated with 10 or 20 ng/ml of IGF-1 for 24, 36, and 48 h and 1.0, 10, or 100 ng/ml of TNF-α for 24 h. The selected conditions for IGF-1 were 20 ng/ml for 24 and 36 h and for TNF-α 100 ng/ml for 24 h.
In Vivo Experiments
C57BL6/J female or male mice, 2 months of age at the start of the experiments, were used. Animal care and treatment were conducted in conformity with the institutional guidelines that are in compliance with national (D.L. n.116, G.U., suppl 40, 18 Febbraio 1992, Circolare No. 8, G.U., 14 Luglio 1994) and international laws and policies (EEC Council Directive 86/609, OJL 358, Dec 1987; U.S. National Institutes of Health Guidelines). Animals were housed in a constant temperature room with a 12-h dark–12-h light cycle and fed a standard diet. AKI was induced in female mice by subcutaneous injection of cisplatin (18 mg/ml) dissolved in sterile 0.9% saline solution. The dose of cisplatin was chosen on the basis of preliminary experiments, and the highest sublethal dose was used.
To study intrarenal localization and perform quantification of MSCs, 2 × 105 cells were labeled with PKH-26 (16) red fluorescence cell linker and then were infused in cisplatin-treated mice, which were sacrificed at 1 day. Before injection, MSCs were preconditioned with IGF-1 (20 ng/ml) (n = 3), GDNF (100 ng/ml) (n = 3), or TNF-α (100 ng/ml) (n = 3) for 24 h. Labeling efficacy of PKH-26-labeled MSCs was >98%. Viability, evaluated by trypan blue exclusion, was >96%.
For renal morphology and function studies, female C57BL6/J mice were divided in three groups and, 1 day after cisplatin administration, received an intravenous (IV) injection as follows: group 1, saline (n = 6–8); group 2, 2 × 105 MSCs derived from bone marrow of male C57BL6/J mice (n = 6–8); group 3, 2 × 105 MSCs preconditioned with IGF-1 (n = 6–8). Before injection, MSCs were incubated for 24 h with IGF-1 (20 ng/ml). Mice were killed at 4 days after cisplatin injection, and blood samples for blood urea nitrogen (BUN) determination were collected before (day 0) and at day 4 after cisplatin administration. Renal function was assessed as BUN in heparinized blood by the Reflotron test (Roche Diagnostics, Indianapolis, IN). BUN levels in control animals averaged 17.4 ± 0.78 mg/dl. Kidneys were taken at day 4 for histological and ultrastructural analysis.
Quantification of MSC Engraftment in Renal Tissue
Kidney samples were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), embedded in 30% sucrose/PBS and in Tissue-Tek OCT Compound (Sakura Finetek, Torrance, CA), and then fresh-frozen in liquid nitrogen. Three-micrometer-thick sections were fixed in acetone (10 min) and incubated with fluorescein isothiocyanate (FITC)-labeled lectin wheat germ agglutinin (WGA; Vector Laboratories, Burlingame, CA), which binds membrane glycoproteins and sialic acid and was used to identify renal structures. Nuclei were stained with 4,6-diamidino-2-phenylindole dihydrochloride hydrate (DAPI). Fifteen sections per mouse were analyzed, and both PKH-26+ MSCs and DAPI+ renal cells were counted in each field. Data were then expressed as number of PKH-26-positive cells per 100,000 renal cells. Stem cell engraftment was also evaluated in heart, lungs, brain, and liver.
Light Microscopy
Fragments of renal cortex were fixed in Dubosq-Brazil (DDK Italia, Milan, Italy) and embedded in paraffin. Sections of 3-μm thickness were stained with hematoxylin and eosin, Masson's trichrome, or periodic acid-Schiff (PAS) reagent (all from BioOptica, Milan, Italy). Luminal hyaline casts and tubular cell degenerative changes (cytoplasmic vacuolization, swelling, cell flattening, PAS-positive droplets, nuclear fragmentation, cell debris), and cell loss (denudation of the tubular basement membrane) were assessed in nonoverlapping fields (up to 20 fields for each mouse) of the entire section taken at high magnification of 400x (high-power field, HPF). Number of casts and tubular profiles showing necrosis were recorded in a single-blind fashion.
Immunofluorescence Analysis
MSCs seeded on glass coverslips at 2.5 × 104 cell/cm2 in 10% FCS medium were switched to 2% FCS plus IGF-1 (20 ng/ml) for 24 h and then fixed in 2% paraformaldehyde plus 4% sucrose and permeabilized with 0.3% Triton X-100. After incubation with blocking solution (2% fetal bovine serum, 2% bovine serum albumin, and 0.2% bovine gelatin in PBS), cells were incubated overnight at 4°C with rat anti-mouse CXCR4 antibody (R&D Systems) followed by the secondary antibody FITC-conjugated goat anti-rat IgG (Jackson Immunoresearch, West Grove, PA) and then with 20 U/ml rhodamine phalloidin (Molecular Probes, Inc., Eugene, OR) for 20 min. Finally, cells were treated with the nuclear cell marker DAPI for 15 min at 37°C. Coverslips were washed, mounted in 1% N-propyl-gallate in 50% glycerol and 0.1 mol/L Tris–HCl, pH 8, and examined using a confocal inverted laser microscope (LSM 510 Meta, Zeiss). Fifteen fields, systematically digitized along the adhesion surface, were acquired using a computer-based image analysis system. The fluorescent area was evaluated by automatic edge detection using built-in specific functions of the software Image J (NIH, Bethesda, MD) and expressed as pixel2 per field analyzed.
To study cytoskeletal architecture, cells were incubated with IGF-1 (20 ng/ml) for 36 h, GDNF (100 ng/ml), or TNF-α (100 ng/ml) for 24 h. Fixed cells were permeabilized, saturated in blocking solution for 30 min, and incubated overnight at 4°C with polyclonal goat antivimentin antibody (Santa Cruz Biotechnology, USA, CA). Then cells were incubated with FITC-conjugated rabbit anti-goat (Jackson Immunoresearch) for 1 h at room temperature (RT). To visualize F-actin filaments, cells were further incubated with 20 U/ml rhodamine phalloidin (Molecular Probes, Inc.) for 20 min; negative control experiments were also performed. At the end of incubation, cells were treated with DAPI, mounted, and examined microscopically.
FACS Analysis
For fluorescence-activated cell sorting (FACS) analysis of CXCR4 surface expression, cell suspensions of MSCs untreated or preconditioned with IGF-1 (20 ng/ml), GDNF (100 ng/ml), or TNF-α (100 ng/ml) (n = 3) for 24 h were incubated with rat anti-mouse CXCR4 (R&D Systems) in blocking solution for 30 min. IgG2b was used as negative control. The incubation of the secondary antibody was performed using FITC-conjugated goat anti-rat IgG (Jackson Immunoresearch) for 1 h at RT, and cells were detected by FACS (Becton Dickinson, Franklin Lakes, NJ).
Western Blot Analysis
Adherent cells were rinsed with cold PBS plus 0.1 mmol/L sodium orthovanadate and lysed in hot sodium dodecyl sulfate (SDS) sample buffer. Whole-cell lysates were separated by SDS polyacrylamide gel electrophoresis and transferred onto nitrocellulose filters (VWR International PBI Srl, Milan, Italy). Blots were blocked with 2% (w/v) ECL Advance blocking agent (ECL™ Advance, GE Healthcare, UK) in TBS-T [25 mmol/L Tris–HCl (pH 7.2), 150 mmol/L NaCl, 0.1% Tween 20] for 2 h at RT. Then filters were incubated with rabbit anti-CXCR4 antibody (1:500; Abcam), which recognises two bands, one at about 39 kDa corresponding to the monomer and one at about 78 kDa corresponding to the dimer (1) or with rabbit anti-actin (1:10,000) antibodies overnight. Finally, blots were incubated with peroxidase-conjugated anti-rabbit antibody (1:40,000; Sigma-Aldrich) to reveal immunoreactive bands using the enhanced chemiluminescence detection system (ECL™ Advance).
Assessment of Cell Viability and Apoptosis
MSCs seeded in triplicates into 12-well plates were exposed to medium alone or IGF-1 20 ng/ml for 24 h and further incubated with 200 μM H2O2 for 24 h. Adherent and floating cells were collected and counted with a cell counter (Digital Bio, Boston, MA). Cell viability was expressed as percentage of propidium iodide (PI; NanoEnTek, Inc., Newton, MA)-negative cells over the total cell number. Adherent and floating cells were analyzed for DNA fragmentation (apoptosis) by terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) assay (Roche, Mannheim, Germany) according to the manufacturer's recommendation. Fluorescence was measured by flow cytometry using a FACSort (Becton Dickinson, Franklin Lakes, NJ).
Measurement of Growth Factors in Cell Supernatants
MSCs were treated or not with IGF-1 for 24 h in serum-free conditions. Afterwards, medium was removed, and cells were washed twice with PBS and cultured in serum free conditions for further 24 h before collecting the supernatants. Supernatant levels of basic fibroblast growth factor (bFGF) and vascular endothelial growth factor (VEGF) were measured using a magnetic bead-based assay (Bio-Plex Pro cytokine assay, Mouse Group II; Bio-Rad, Hercules, CA, USA). Multianalyte profiling was performed by using Luminex xMAP technology on the Bioplex 200 Suspension Array System (Bio-Rad, Hercules, CA, USA). Analyses were performed according to the manufacturers' protocols, and data were processed with Bio-Plex Manager 5.0 software (Bio-Rad, Hercules, CA, USA). Data analysis was done with five-parametric-curve fitting.
Levels of IGF-1 and transforming growth factor-β1 (TGF-β1) were measured in the culture supernatant by using, respectively, the Mouse/Rat IGF-1 Quantikine ELISA Kit and the Second Generation Mouse/Rat/Porcine/ Canine TGF-β1 Quantikine ELISA Kit (R&D Systems, Inc., Minneapolis, MN, USA). Hepatocyte growth factor (HGF) was detected using the Mouse HGF ELISA Kit for cell culture supernatant (RayBiotech, Norcross, GA, USA). All samples were coded for a blinded analysis, and each sample concentration was determined in duplicate. The results were expressed in pg/ml/105 cells.
Statistical Analyses
The results are expressed as means ± SEM. Statistical analysis was performed using ANOVA followed by Tukey Cicchetti test for multiple comparisons or Bonferroni post hoc analysis. Statistical significance level was defined as p < 0.05 or as indicated.
Results
IGF-1, GDNF, or TNF-α Induces Morphological Cytoskeletal Changes of MSCs
Cellular migration requires morphological changes, wherein cells are structurally reorganized and cytoskeletal filaments adopt a well-ordered, parallel orientation (22,37). Thus, we studied the effects of IGF-1, GDNF, or TNF-α on MSC cytoskeleton by analyzing the filamentous pattern and distribution of F-actin and vimentin. In normal conditions, a large number of cells showed an unpatterned orientation of cytoskeleton with filaments to be organized in independent directions. In the presence of IGF-1, GDNF, or TNF-α, most of the cells displayed parallel-oriented filaments organized along the axis of the cells (Fig. 1). These structural changes in response to the above factors may be indicative of an architectural remodeling, contributory to the mechanical needs of migration.
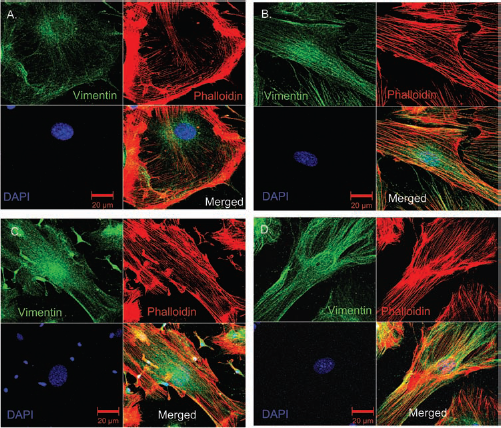
IGF-1, GDNF, and TNF-α treatment induces changes on MSC cytoskeletal proteins. Insulin-like growth factor-1 (IGF-1; 20 ng/ml), glial cell-derived growth factor (GDNF) (100 ng/ml), and tumor necrosis factor (TNF)-α (100 ng/ml) induce cytoskeletal rearrangements, shown by phalloidin staining (red) and immunocytochemical detection of vimentin (green). Representative images of mesenchymal stem cell (MSC) cytoskeletal organization under control conditions (A) or after incubation with IGF-1 (B), GDNF (C), and TNF-α (D). In untreated cells, filaments are organized in independent directions, while after the treatment they display a parallel orientation.
IGF-1, GDNF, or TNF-α Increase the Migration Capacity of MSCs
In order to spot the most promigratory agents we used two different in vitro approaches. Firstly, we applied a scrape-healing assay where cells were scratched and MSC healing capacity was quantified (Fig. 2A, B). Results showed that treatment with IGF-1, GDNF, or TNF-α enhanced migration of the surrounding cells towards the injury, reducing the surface area of the scratch, which after 20 h was almost closed (Fig. 2A). In contrast, untreated MSCs displayed a low migratory activity in response to the injury and after 20 h scratch was still visible and distinct. To confirm that our results were due to MSC migration and not proliferation, we counted MSCs at the end of scrape-healing experiments and no differences were observed among the groups (MSC control: 2.45 ± 0.09 × 104, MSC/IGF: 2.53 ± 0.13 × 104, MSC/GDNF: 245 ± 0.05 × 104, MSC/TNF-α: 2.52 ± 0.11 × 104 cells/cm2) (n = 3).

Preconditioning with IGF-1, GDNF, or TNF-α enhances MSC migration capacity. (A) Scrape-healing assay of MSCs under control conditions or after treatment with IGF-1 (20 ng/ml), GDNF (100 ng/ml), or TNF-α (100 ng/ml). After scratching, the surrounding cells migrated into the scraped zone. Representative images were taken 20 h after scratching. (B) Quantification of MSC healing capacity. Data are mean ± SEM. ∗p < 0.05 versus untreated MSCs. (C) Transmigration of untreated, IGF-, GDNF-, or TNF-α-treated MSCs towards control or cisplatin-injured PTECs. Data are mean ± SEM. ∗p < 0.05 versus untreated MSCs in coculture with cisPt/PTECs. #p < 0.05 versus the corresponding MSCs in coculture with control PTECs. PTECs, proximal tubular epithelial cells; CisPt, cisplatin.
Afterwards we applied an in vitro injury–migration model, based on a transwell system consisting of murine MSCs cocultured with cisplatin-injured proximal tubular epithelial cells (PTECs) (Fig. 2C). Migration of MSCs from the upper chamber across the membrane towards the cisplatin-damaged tubular cells was significantly higher than that observed when MSCs were in coculture with control PTECs (Fig. 2C). Preconditioning of MSCs with IGF-1, GDNF, or TNF-α amplified the migration response of the cells to cisplatin-injured PTECs as determined by a significant increase in the number of migrating cells found in the lower site of transfilter (Fig. 2C). Preconditioned cells that were cocultured with untreated PTECs did not exhibit significant migration activity (Fig. 2C).
Preconditioning with IGF-1 Enhances MSC Engraftment In Vivo
We studied whether MSC preconditioning with IGF-1, GDNF, or TNF-α before their in vivo infusion might influence MSC engraftment in damaged tissue of mice with acute kidney injury. Female C57BL6/J mice were injected with cisplatin, an antitumor drug whose clinical use is accompanied by high incidence of nephrotoxicity, associated with renal dysfunction and renal damage with extended structural alterations (29,40). One day after cisplatin injection, mice were systemically infused with MSCs, and 24 h later cells were monitored in the injured kidney. Preconditioning of MSCs with IGF-1 before infusion significantly increased the migration of PKH26-labeled cells into the cisplatin-injured kidney (34.8 ± 3.8 cells/100,000 renal cells), as compared to untreated MSCs (16.1 ± 1.4 cells/100,000 renal cells) (Fig. 3A), while GDNF (23.9 ± 0.5 cells/100,000 renal cells) or TNF-α (18.1 ± 2.4 cells/100,000 renal cells) had lower or no effect on engraftment ability of MSCs. Engrafted cells in kidneys of mice with AKI were predominantly found in the peritubular areas (Fig. 3B) (60 ± 10% of total engrafted cells), and rarely in the context of tubular epithelium (0.8 ± 0.4%), while a number of PKH26+ cells were observed in glomeruli (39.2 ± 10%), which rapidly disappeared with time.

Effect of IGF-1 preconditioning on MSC engraftment in kidneys of cisplatin-treated mice after 24 h. (A) Quantification of PKH26-labeled untreated (MSC) or IGF-1-treated (MSC/IGF) MSCs in cisplatin-injured renal tissue 24 h after injection. Data are mean ± SEM. ∗p < 0.05 versus MSCs. (B) PKH-26-labeled cells (red fluorescence, arrows) were localized in the peritubular areas. Sections were stained with DAPI (nuclear staining, blue) followed by Wheat Germ Agglutinin (WGA) Lectin (green).
Preconditioning with IGF-1 Enhances MSC-Mediated Protective Effect on Renal Structure and Function
As IGF-1 preconditioning proved to be the most effective strategy for facilitating MSC engraftment in damaged tissue, we further examined the capacity of IGF-1-treated cells to restore renal structure and function after acute injury. Mice injected with cisplatin developed renal function impairment, characterized by high serum level of BUN, between days 4 and 7 after drug administration (29). Intravenous injection of 2 × 105 MSCs obtained from the BM of male mice into syngeneic female mice 1 day after cisplatin treatment protected mice from renal function deterioration, as shown by significantly lower BUN values (65.52 ± 14.9 mg/dl) at 4 days, with respect to cisplatin-treated mice that received saline (104.16 ± 9.22 mg/dl) (Fig. 4A). Injection of IGF-1-treated MSCs into cisplatin-injured mice resulted in a further BUN decrease (29.9 ± 6.42 mg/dl, p < 0.05) as compared to both saline-treated and MSC-treated mice (Fig. 4A).

IGF-1-treated MSCs exert a strong renal protective effect on mice with cisplatin-induced acute kidney injury (AKI). (A) Blood urea nitrogen (BUN) was evaluated before (basal) and 4 days after cisplatin treatment in mice that received an intravenous injection (IV) of saline, unstimulated MSCs (2 × 105 cells) or IGF-1-treated MSCs (MSC/IGF, 2 × 105 cells). Data are means ± SEM. ∗p < 0.05 versus MSCs, #p < 0.05 versus basal, #p < 0.05 versus saline. (B) Histological examination of kidney sections of C57BL6/J cisplatin-treated mice, given (IV) saline, MSCs, or IGF-1-treated MSCs at 4 days. Representative images of the kidney of cisplatin-treated mouse receiving saline showing tubular epithelial cell degeneration and necrosis (arrows) and cast formation (∗). Treatment with MSCs resulted in less severe tubular damage with occasional luminal casts. Tubular lesions in cisplatin mice receiving IGF-1-preconditioned MSCs were almost absent. (C) Quantification of engrafted PKH26-positive MSCs in cisplatin-injured renal tissue at 4 days. Data are means ± SEM. ∗p < 0.05 versus MSCs.
We next investigated whether renal function improvement was related to preservation of tubular structures. By light microscopy, normal kidneys showed no structural alterations, while the kidneys of cisplatin-treated mice given saline displayed significant tubular lesions at 4 days (p < 0.05 vs. normal), consisting of loss of brush border, flattening and loss of the tubular epithelium, nuclear fragmentation, and hyaline casts (Fig. 4B). Renal tissue of mice receiving untreated MSCs showed a significant reduction in cast formation and tubular necrosis in respect to saline-treated mice (Fig. 4B and Table 1). Remarkably, mice injected with IGF-1-treated MSCs showed further reduction in tubular necrosis and overall only minor ultrastructural tubular changes (Fig. 4B and Table. 1).
Effects of MSCs on Renal Histology at 4 Days

Quantification of casts and necrotic tubuli, as numbers per high-power field (HPF), in cisplatin-treated mice at 4 days receiving saline, mesenchymal stem cells (MSCs), or insulin-like growth factor-1 (IGF-1)-treated MSCs. Normal kidneys exhibited no structural alterations. Data are mean scores ± SEM.
p < 0.001
p < 0.005 versus saline
p < 0.05 versus MSC.
Concomitantly, the presence of IGF-1-treated MSCs at 4 days was still higher in the injured kidney (4.2-fold, p < 0.05) than untreated MSCs (Fig. 4C). Furthermore, between 1 and 4 days a dramatic decline in the number of engrafted cells occurred, wherein only 6.8% of the initial number of untreated cells was still present in renal tissue. In contrast, the percentage of remaining IGF-1-treated cells in the same time course was 13.0% of the initial number, which may imply a possible prosurvival effect of IGF-1 on exogenously infused MSCs. No PKH26+ cells were detected in heart, lungs and brain, while few cells were found in the liver (2.4 ± 0.7 PKH-26+ cells/100,000 liver cells).
IGF-1 Improves MSC Survival In Vitro and Stimulates IGF-1 Production
To examine whether IGF-1 exerts prosurvival effects on MSCs, we cultured cells preconditioned with IGF-1 or not, at the presence of H2O2 (200 μM, 24 h) (Fig 5A). Our results showed that H2O2 significantly reduced MSC viability in respect to untreated cells (20.0 ± 2.2% vs. 97.5 ± 0.3%, p < 0.05), while IGF-1 treatment significantly limited MSC susceptibility to H2O2 (64.9 ± 0.5% vs. 20.0 ± 2.2%, p < 0.05). Under the same conditions apoptosis of MSCs was dramatically reduced in IGF-1-treated cultures (Fig. 5B).
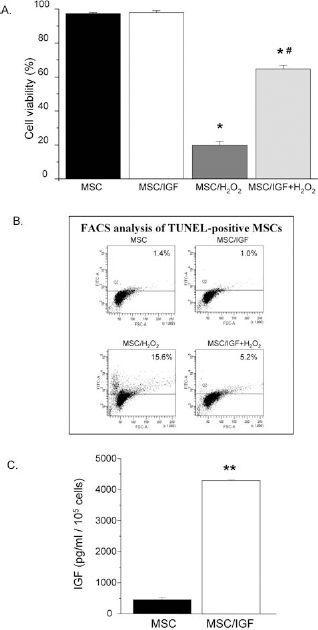
IGF-1 improves MSC survival in vitro and stimulates IGF-1 production. (A) MSCs were exposed to medium alone (MSC), IGF-1 (MSC/IGF), H2O2 (MSC/H2O2), or IGF-1 and H2O2 (MSC/IGF + H2O2). IGF-1 treatment significantly reduced MSC susceptibility to H2O2. Results are the mean ± SEM of five independent experiments. ∗p < 0.05 versus MSC and MSC/IGF; #p < 0.05 versus MSC/H2O2. (B) Representative graphs of the percentage of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive MSCs (upper quadrant) analyzed by fluorescence-activated cell sorting (FACS). IGF-1 dramatically limited apoptotic death of MSCs exposed to H2O2. One of two representative experiments is shown. (C) Incubation of MSCs with IGF-1 showed a marked increase in IGF-1 secretion as compared to control cells (MSC). Results are the mean ± SEM of four independent experiments. ∗∗p < 0.000005 versus MSC.
Since it has been previously documented (16,41,43) that MSCs exert their beneficial effects via paracrine production of antiapoptotic, mitogenic, and vasculotrophic factors, we investigated the effect of IGF-1 on MSC ability to produce IGF-1, TGF-β1, HGF, bFGF, VEGF by multianalyte profiling assay. Our results showed that MSCs constitutively release considerable amounts of IGF-1, TGF-β1, HGF, bFGF, and VEGF (470.25 ± 51.09, 42.30 ± 3.51, 576.57 ± 88.95, 100.66 ± 23.68, and 2,293.04 ± 64.00 pg/ml/105 cells, respectively). When MSCs were exposed to IGF-1 a 10-fold increase (4,302.07 ± 21.58 pg/ml/105 cells) in the production of IGF-1 was observed (Fig. 5C).
IGF-1 Induces Changes in CXCR4 Cellular Expression
In order to investigate the mechanisms underlying the enhanced renal migration and protective effect of MSCs preconditioned with IGF-1, we focused on CXCR4 that plays a pivotal role in the migration and engraftment of BM-MSCs (6,12,35). Therefore, we examined the surface expression of CXCR4 in response to IGF-1 by FACS (Fig. 6A and Table 2). Results showed that IGF-1 treatment induced a twofold increase of CXCR4 expression on MSC surface (Table 2). At variance, GDNF (3.2%) and TNF-α (3.5%) do not exhibit any significant increase on CXRC4 surface expression as compared to corresponding MSC controls (2.5% and 2.6%, respectively).
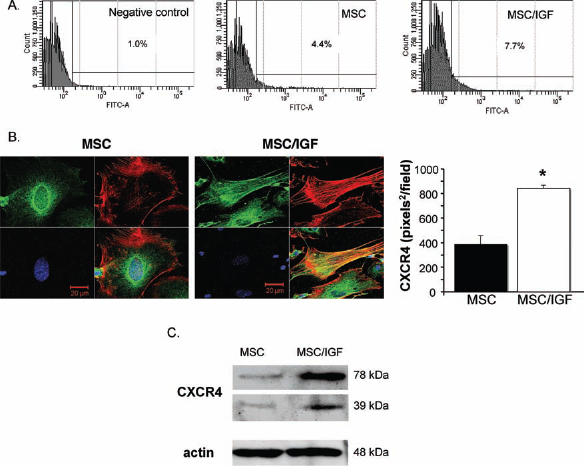
IGF-1 preconditioning of MSCs increases CXCR4 expression. (A) Representative fluorescence histograms showing the C-X-C chemokine receptor type 4 (CXCR4) expression in untreated and IGF-1-treated MSCs by FACS. (B) Representative immunofluorescence images of intracellular CXCR4 (green) expression and F-actin (red) in untreated MSCs (left) or treated with IGF-1 (20 ng/ml) for 24 h (central). Quantification of intracellular CXCR4 in untreated or IGF-1-treated MSCs using a computer based image analysis system (right). Data are mean ± SEM. ∗p < 0.05 versus untreated MSCs. (C) Western blot analysis of CXCR4 expression in MSCs treated or not with IGF-1 for 24 h. Two bands were recognized at about 39 kDa (monomer) and 78 kDa (dimer). Control loading is shown by actin.
Expression of CXCR4 by FACS Analysis

Data are mean ± SEM of five independent experiments. CXCR4, C-X-C chemokine receptor type 4; FACS, fluorescence activated cell sorting.
p < 0.05 versus MSC.
Consistently, evaluation of CXCR4 in MSCs by a computer-based image analysis system showed a significant increase in the intracellular granular staining of CXCR4 in IGF-1-treated cells (twofold, p < 0.05), in comparison to untreated cells (Fig. 6B). Furthermore, Western blotting experiments showed that IGF-1 stimulates the expression of total CXCR4 (Fig. 6C).
To define the biological significance of IGF-1-mediated CXCR4 upregulation, we performed scrape-healing experiments in the presence of a neutralizing antibody. The functional blocking of CXCR4 totally abrogated the promigratory effect of IGF-1 on MSCs (Fig. 7A). Additionally, motility parameters as total distance (Fig. 7B), straight distance (Fig. 7C), and velocity (Fig. 7D), which were significantly increased in cells exposed to IGF-1, normalized back to control values by CXCR4 antibody, indicating a key role for CXCR4 on IGF-1-induced migration.
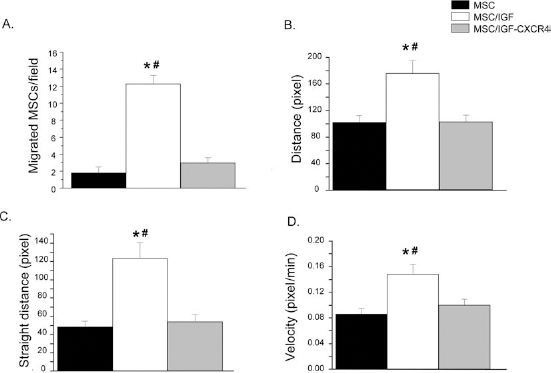
Role of CXCR4 in IGF-1 promigratory effects on MSCs. (A) Quantification of cell migration in untreated MSCs (black) or treated with IGF-1 (white) or IGF-1 plus anti-CXCR4 neutralizing antibody (gray) 20 h after scratching. (B–D) Analysis of different parameters of MSC motility 20 h after scratching in untreated MSCs (black) or treated with IGF-1 (white) or IGF-1 plus anti-CXCR4 neutralizing antibody (gray). (B) Distance, (C) straight distance, and (D) velocity. Data are mean ± SEM of two independent experiments. ∗p < 0.05 versus untreated MSCs, #p < 0.05 versus MSCs treated with IGF-1 plus CXCR4 Ab.
Discussion
Presently, the delivery of MSCs via systemic infusion is the predominant route being used in the majority of the trials since local transplantation not only is it highly invasive but also seems to involve excessive cell death early after local administration, mainly due to lack of nutrients and oxygen (32). However, with evidence for massive entrapment of stem cells in the lungs and in capillary beds of different tissues, the need of developing new strategies to enhance cell homing to target tissues emerges as a prerequisite for the success of MSC-based systemic therapies. Here we documented a novel approach to optimize MSC engraftment efficiency and increase their therapeutic effects, based on a preconditioning with IGF-1 before systemic infusion. The most promigratory compounds were initially chosen through an in vitro injury–migration model consisting of MSCs and cisplatin-damaged tubular cells and by applying a scrape-healing setting. From our in vitro studies, we have found that preconditioning of MSCs by IGF-1, GDNF, or TNF-α induced a marked cytoskeletal rearrangement, enhanced migration towards injured tubular cells, and accelerated migration in the scrape-healing assay. In vivo, however, IGF-1 showed higher ability to improve MSC engraftment in the cisplatin-injured kidney. Preconditioning with IGF-1 also resulted in remarkable amplification of the protective effect of MSCs to such an extent that BUN values were comparable to the normal values obtained from healthy animals. Similarly to the effect on renal function, MSCs treated with IGF-1 completely abrogated renal structure damage, implying a coordinated functional and structural protective effect of preconditioned cells in cisplatin-induced AKI.
Several studies have proposed the axis stromal cell derived factor (SDF-1)/CXCR4 as a pivotal mediator of migration and engraftment of MSCs (6,12,35). Here, we observed that CXCR4 was constitutively expressed at the cell membrane and upregulated by IGF-1 treatment. Interestingly, IGF-1 induced a twofold increase, both in the number of cells expressing CXCR4 and in the number of engrafted cells, indicating a correlation between CXCR4 surface expression and engraftment. Furthermore, evaluation of the intracellular storage and total CXCR4 in IGF-treated cells showed a significant increase in respect to untreated cells. The functional role of CXCR4 upregulation in response to IGF-1 was documented by CXCR4 blocking which totally abrogated the promigratory effect of IGF-1 on MSCs and affected crucial motility parameters as distance and velocity. These findings indicate that CXCR4 plays a key role in migration of IGF-1-treated MSCs in vitro and could well explain the increased engraftment of these cells in vivo. In support of this hypothesis, we also detected an augmented expression of SDF-1, the ligand of CXCR4, in renal damaged tissue of cisplatin mice (data not shown).
Although we observed only a small increase in the percentage of the cells expressing surface CXCR4, this difference was of biological significance and able to affect migration capacity of MSCs, consistent with previous studies showing that a slight increase in the percentage CXCR4-expressing MSCs induced by short-term hypoxia resulted in significant improvement of migration capacity (15). On the other hand, the increased intracellular expression of CXCR4 might serve as a receptor reservoir (19,48), which can be potentially induced to cell surface in response to IGF-1 stimulation. Unlike IGF-1, GDNF and TNF-α did not exhibit any effect on CXCR4 expression. This difference in CXCR4 expression between IGF-1 preconditioning and the other factors can well justify the fact that only IGF-1 displayed significant effects on MSC engraftment into the injured tissue.
Previous studies showed that MSC exert beneficial effects on tubular cell repair in acute kidney injury by producing the mitogenic and prosurvival factor IGF-1. Knocking down IGF-1 expression in MSCs decreased tubular cell proliferation, increased apoptosis, and limited their protective effect on renal function (16). Here we documented that IGF-1 preconditioning strongly enhanced the reno-protective potential of infused MSCs, as well as induced a dramatic secretion of IGF-1, suggesting that IGF-1-treated MSCs exert their regenerative effects on tubular damage by means of an excessive release of IGF-1 at the sites of the injury.
The relative paucity of MSCs in the renal tissue in the face of their remarkable protective effects observed herein is consistent with previous findings in animals with AKI, showing that injected MSCs, transiently localized at early time points, in peritubular areas, and disappeared after 24–72 h in ischemic rat kidney (41). The frequency of IGF-1-preconditioned cells however was increased in the injured kidney at 1 day and still remained higher at 4 day as compared to untreated MSCs. The fact that IGF-1 preconditioning delays the rate of decline of engrafted cells suggests a beneficial effect of IGF-1 not only on the migration potentials of these cells but also on their longevity in the target damaged tissue. This delay in depletion of IGF-1-treated cells from injured kidney might be due to prosurvival signaling stimulated by IGF-1. Consistently, in vitro experiments showed that IGF-1-treated MSCs were less sensitive to the oxidative damage induced by H2O2. Altogether these data potentially suggest that IGF-1 improves the resistance of MSCs against the proapoptotic microenvironment of the injured renal tissue.
The therapeutic impact and feasibility of the current approach was evaluated by using a murine cisplatin-induced AKI model, not only as it is a well-defined and highly reproducible model (29–31) but also because the wide use of cisplatin as a chemotherapeutic agent potentially renders the current approach remarkably constructive and therapeutically relevant. In fact, a relevant clinical trial using ex vivo expanded third party mesenchymal stem cells to repair renal function in cisplatin-induced AKI in patients with solid organ cancers is already approved by regulatory authorities. Considering the beneficial effect of MSCs in cisplatin-induced AKI, we chose to use a relatively high dose of cisplatin, in order to bring forth the effect of IGF-1 preconditioning. Interestingly, even with an uncommonly high dose of cisplatin, which was able to induce severe functional and structural renal alterations, IGF-1-treated MSCs were highly protective and capable to abrogate almost completely the detrimental effects of cisplatin.
In comparison with other strategies aiming to improve efficacy of stem cells, in particular genetic modification, ex vivo preconditioning with IGF-1 presents diverse advantages in terms of clinical feasibility. First, IGF-1 can be administered to MSCs with negligible in vitro manipulations, avoiding extended passaging and persisting incubations, which may interfere with cell quality. Second, IGF-1 can be entirely removed before injection without loss of its beneficial properties, rendering the risk of side effects particularly unlikely.
Here, we report a “multum in parvo” therapeutic approach, which amplifies the beneficial effect of MSCs in tissue injury, through an effortless preconditioning with IGF-1. Our results showed that preconditioning of MSCs with IGF-1 before infusion improves their migration capacity and restores normal renal function after AKI. These data represent a new therapeutic paradigm, suggesting that improving MSC migration ability could increase therapeutically relevant effects. The enhanced delivery to specific tissues could increase the efficiency of cell therapy and reduce the number of infused cells, potentially limiting the complications and the cost of developing a therapeutic product. In as much as MSCs are already used safely in clinical studies, it is plausible that this novel therapeutic paradigm may be implemented rapidly in the clinical setting or facilitates the development of novel cell-based therapies for various diseases and damages.
Footnotes
Acknowledgments
We thank Daniela Corna, Cinzia Rota, Daniela Rottoli, Lorena Longaretti, and Chiara Ardeleani for excellent technical assistance. We also thank Leda Vassilopoullou and Joanna Johnson for manuscript corrections. This study was supported by Research Training Network “Developing a stem cell based therapy to replace nephrons lost through reflux nephropathy” (http://www.kidstem.org) funded by the European Community as part of the Framework program 6 (FP6 036097-2) and by Juvenile Diabetes Research Foundation (JDRF) International. Part of the research leading to these results has also received funding from the European Community under the European Community's Seventh Framework Programme (FP7/2007-2013), grant number 223007, STAR-TREK project. Valentina Benedetti is recipient of a fellowship from “Fondazione Aiuti per la Ricerca sulle Malattie Rare” (ARMR), Bergamo, Italy. The authors declare no potential conflicts of interest.