
Abstract
The limited availability of liver donors and recent progress in cell therapy technologies has centered interest on cell transplantation as a therapeutic alternative to orthotopic liver transplant for restoring liver function. Following transplant by intraportal perfusion, the main obstacle to cell integration in the parenchyma is the endothelial barrier. Transplanted cells form emboli in the portal branches, inducing ischemia and reperfusion injury, which cause disruption of endothelial impermeability and activate the immune system. Approximately 95% of transplanted cells fail to implant and die within hours by anoikis or are destroyed by the host immune system. Intravascular perfusion of Bordetella pertussis toxin (PTx) blocks endothelial Gi proteins and acts as a reversible inducer of actin cytoskeleton reorganization, leading to interruption of cell confluence in vitro and increased vascular permeability in vivo. PTx treatment of the murine portal vascular tree 2 h before intraportal perfusion of embryonic stem cells facilitated rapid cell engraftment. By 2 h postperfusion, the number of implanted cells in treated mice was more than fivefold greater than in untreated controls, a difference that was maintained to at least 30 days posttransplant. We conclude that prior to cell transplant, PTx blockade of the Gi protein pathway in liver endothelium promotes rapid, efficient cell implantation in liver parenchyma, and blocks chemokine receptor signaling, an essential step in early activation of the immune system.
Introduction
The best therapeutic alternative in severe liver failure, both acute and chronic, is currently an orthotopic liver transplant (33). Donor availability nonetheless falls far short of the increasing demand, whereas the number of potential donors remains stable, resulting in delays and increased mortality of waiting list patients (1,2). Limited organ availability and recent developments in cell therapy technology have focused interest on cell transplant technology as a therapeutic alternative for restoring liver mass and function (57). The special vascular structure of the liver makes this organ an excellent candidate for cell transplant. As liver sinusoids (capillaries) lack a basement membrane and an intraportal cell perfusion system, endothelial cells are the only barrier between the perfused cells and the parenchyma into which they are to be integrated (40). Among the advantages of cell transplant compared to orthotopic liver transplant are the reduction in surgical complexity, rapid patient recovery, lower costs, and that, at difference from the whole organ, cells can be cryopreserved for use in emergency situations such as fulminant hepatitis (57,58).
In murine models of hepatocyte transplant, good liver repopulation ratios can be obtained, with restoration of organ function (5,43,61). These results have not been reproduced satisfactorily in humans (50,57), although partial improvement has been achieved in patients with metabolic disorders, complete correction of disease has yet to be reported (9,15,25,39,47,50). The principal constraints to the introduction of cell transplant in clinical practice are a shortage of appropriate cells for transplant and their limited implantation efficiency (12). Major advances have been made in optimizing protocols to obtain, transport, store, and perfuse cells (44,58), but unsatisfactory implantation results limit overall transplant efficiency (63).
After intraportal hepatocyte transplant, approximately 95% of transplanted cells die within the first 24 h by anoikis, a form of apoptosis triggered by lack of survival signals induced by adhesion molecule binding to the extracellular matrix (48,56), or by cell destruction by the host immune system (23). Low cell transplant efficiency is not unique to hepatocytes, but is also observed in other human cell transplant models (30). Conditioning treatments that enhance cell implant have shown promising results in rodent models (35). Such therapies increase vascular permeability and allow transplanted cells to move from the circulation to the perisinusoidal space (space of Disse) and integrate into the tissue (34). Most of these implantation enhancers nonetheless have limited usefulness, as they are toxic or slow to take effect (28,29,63).
We previously reported an in vitro method for evaluating potential engraftment enhancers, based on their ability to disrupt endothelial cell adhesion. In an in vivo model, this would allow an increase in vascular permeability sufficient to permit transplanted cells to pass from the circulation to the perisinusoidal space (3). Using this approach, we identified B. pertussis toxin (PTx) as a possible potentiator of cell transplant. PTx is secreted by the bacterium during infection and acts by catalyzing ADP ribosylation of Gi proteins, which prevents Gi protein interaction with cell membrane receptors and interferes with intracellular communication (27). Among other effects, inactivation of Gi proteins block signaling through chemokine, histamine, and vasoactive amine receptors involved in regulating initiation of the inflammatory response and vasoregulation (10).
Materials and Methods
Cells and Cell Culture
Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cord by incubating vessel lumen with collagenase H (0.5 mg/ml; Sigma-Aldrich, St. Louis, MO) in 199 medium, as described previously (54), and cultured on 0.1% gelatin-coated (Sigma) plasticware in endothelial cell (EC) medium [199 medium with 10 mM HEPES, 20% fetal calf serum (both from Invitrogen, Carlsbad, CA), 100 μg/ml heparin, and 100 μg/ml endothelial cell growth supplement (both from Sigma)] (54). Institutional and national guidelines for obtaining and manipulating human biological derivatives were followed and protocols were approved by the institutional review board of the Hospital 12 de Octubre.
Mouse liver endothelial cells were obtained by collagenase digestion of liver, purified on an Optiprep gradient (both from Sigma) (14,20), and cultured on 0.1% gelatin-coated petri dishes in EC medium. HUVEC phenotype was determined by fluorescence microscopy using fluorescein isothiocyanate (FITC)-labeled Ulex europaeus lectin (Sigma) and FITC-anti-CD31 (Invitrogen). Purity of EC in cultures was always ≥ 98%. The phenotype of mouse liver endothelial cells was determined by fluorescence microscopy using FITC-Griffonia simplicifolia lectin (Sigma) (6,38); purity of these cells in culture was always ≥ 95%.
For intracellular calcium evaluation, HUVECs were cultured on gelatin-coated coverslip chambers (Nunc, Roskilde, Denmark) for 24 h before treatment (1 h) with 0.5 μg/ml PTx in culture medium. Cells were then washed three times, new medium added, and incubated (30 min, 37°C). Medium was removed and cells treated (20 min) with 5 μM Calcium Green1 (Invitrogen) in PBS; cells were washed twice, new medium added, and incubated (37°C, 5% CO2) until use in experiments (30–90 min). Experiments were performed on a 37°C chamber coupled to a Radiance 2000 MP confocal/multiphoton microscopy system (Bio-Rad, Hercules, CA).
For embryonic stem (ES) cells, we used the B8-6C3 clone (A. Serrano and M. Torres, unpublished data) obtained by gene trapping (59) from R1 murine ES cells; ES cells were cultured as described previously (59). In heterozygosis, these cells express a mutated variant of the alkaline ceramidase gene (Acer3). The mutation eliminates the C-terminal half of the Acer-3 protein (from amino acid 148 to the end) and replaces it with a DNA linker encoding 13 amino acids and cDNA encoding amino acids 22 to 510 of human placental alkaline phosphatase (hPAP). Acer-3 mRNA is expressed ubiquitously, with highest levels in placenta and liver (37); the resulting protein, Acer3-hPAP, shows the tissue and subcellular expression pattern of Acer-3 and enzyme activity of hPAP. Overexpression of hPAP in ES cells has no deleterious effects; transgenic mice ubiquitously expressing hPAP as a reporter show no developmental or viability defects (55).
PTx Treatment of Confluent Cultures
Medium was replaced with fresh medium alone (negative control) or containing PTx (0.1 μg/ml; Sigma). Cultures were fixed at distinct intervals and the cell-free culture surface quantified by image analysis. To evaluate recovery of cytoskeletal reorganization after PTx treatment, HUVECs were seeded in six-well dishes (triplicates for each condition). Confluent cultures were PTx treated as above (1 h), medium removed, cells washed three times with medium 199, and new complete medium added. For controls, PTx was maintained in culture. At 1, 3, 6, 9, and 24 h after initiation of treatment, cultures were fixed (4% formaldehyde) and cell surface (μm2) quantified by image analysis.
Stem Cell Endothelium Adhesion Assay
ES cell adhesion to a PTx-treated confluent EC monolayer was tested as described previously (3) and the relative number of adhered ES cells was quantified by determining alkaline phosphatase (AP) activity using the substrate p-nitrophenyl phosphate after 90-min incubation (3).
Microscopy and Image Processing and Analysis
Cell cultures were fixed with 4% formaldehyde in PBS. HUVECs were stained with FITC-labeled U. europaeus lectin (10 μg/ml, 30 min) in PBS Ca-Mg (supplemented with 1 mM each Ca2+ and Mg2+) and washed three times with the same buffer (for all stainings); mouse endothelial cells were stained with FITC-G. simplicifolia lectin (10 μg/ml) in PBS Ca-Mg. For phalloidin staining, fixed cells were permeabilized with PBS-0.1% Triton X-100 (5 min), and stained with Alexa-488 phalloidin (5 U/ml, 30 min; Invitrogen).
For confocal microscopy of cytoskeleton and of intracellular calcium, we used an IX70 inverted microscope with a 60 PlanApo NA 1.4 objective (Olympus, Tokyo, Japan), coupled to a Radiance 2000 MP microscope (Bio-Rad). Excitation light was 488 nm (argon laser) and images were collected with a 510–560 nm emission filter. Intracellular calcium was measured in 30 time-lapse images captured every 3 s, beginning at 10 s from addition of chemokine (C-C motif) ligand 2 (CCL2; PeproTech, Rocky Hill, NJ; 5 nM final concentration). Relative fluorescence intensity was quantified with ImageJ software.
To evaluate the cultured cell area, cultures were fixed and permeabilized as above, stained with 0.5% Evans blue in PBS (20 min), and viewed on an Olympus IX71 fluorescence inverted microscope using Texas red excitation/emission filters. Five random images were taken for each condition and culture, to allow quantification of at least 400 cells per point.
For confocal microscopy of tissue sections, we used an LSM510 microscope with 10 and 20 PlanApo objectives (Zeiss, Jena, Germany); Cy2 fluorescence was collected between 500 and 550 nm after excitation with the 488-nm line of an argon laser. Cy3 fluorescence was collected between 570 and 683 nm after excitation with a diode laser (561 nm).
Histology and Histochemistry
Mice were sacrificed, livers were extracted immediately and perfused intraportally twice with 20 ml PBS, fixed in 50 ml 4% formaldehyde in PBS (2 h, 4°C), then washed three times with 100 ml PBS (4°C). Sections (50 or 100 μm) were prepared for histology using a VT1200S vibratome (Leica, Mannheim, Germany).
ES cells have strong AP activity (51), much higher than in hepatocytes, and can be distinguished from other cell types by brief treatment (5 min) with nitro blue tetrazolium (BCIP/NBT tablets, Roche, Basel, Switzerland) and 0.1 mM levamisole (Sigma) to inactivate liver AP (49). Implantation was evaluated in three 100μm liver sections per mouse. Three images were captured for each section in an Olympus IX71 inverted microscope with a Nikon DXM 1200 digital camera; images were analyzed and quantified with ImageTool (UTHSC, San Antonio, TX), Adobe Photoshop, and Microsoft Excel. We quantified the percentage of the section surface showing strong AP activity and determined cell implantation rate after 2 h in each mouse as the mean percentage of the AP-positive surface in the nine images. As cells were dispersed throughout the tissue at 7 and 30 days posttransplant, we evaluated long-term implantation by counting the number of cells in each image and adjusting cell number to tissue volume.
Mice
Cell transplant assays were performed using male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME). All experimental protocols were approved by the Hospital Universitario 12 de Octubre Laboratory Animal Care and Use Committee and complied with national and European Union guidelines.
Antibodies
For immunofluorescence of liver sections, we used monoclonal mouse anti-hPAP (clone 8B6, Sigma) and rat anti-mouse CD105 (BioLegend, San Diego, CA) antibodies. Secondary antibodies were Cy2-conjugated goat anti-mouse and Cy3-goat anti-rat IgG (both from Jackson ImmunoResearch, West Grove, PA), used according to manufacturer's recommendations.
Drugs
Following anesthesia with inhaled isoflurane (Forane; Abbott, Berkshire, UK), 12 mice received intrasplenic injections of 100 μl PTx in PBS (5 μg/kg body weight) and 12 received PBS alone. After 2 h, mice were reanesthetized and received a slow intraportal injection of 200 μl DMEM containing ES cells (2 × 106/ml), the abdominal cavity was sutured, and mice were reanimated. For short-term evaluation, mice were sacrificed after 2 h, and livers were removed and fixed as above. Six additional mice (three control, three PTx treated) were sacrificed at 24 h posttransplant and another 12 at 7 and 30 days posttransplant (three control, three PTx treated each). Mice evaluated at 7 and 30 days were treated 24 h posttransplant with an intraperitoneal (IP) bolus of prednisone (2 mg/kg, single dose) and with tacrolimus (2 mg/kg/day, IP) until 1 day before sacrifice.
Visualization of PTx Effect on Hepatic Blood Vessels
PTx or PBS was administered by intraportal injection and mice were sacrificed after 2 h. Livers were processed and sections (50 μm) stained with FITC-G. simplicifolia lectin as above. For each photograph, seven images were taken using bright field illumination with the same x–y coordinates, separated by 7 μm on the z axis. Photographs stacked using ImageJ were deconvolved with Huygens Essential (Scientific Volume Imaging, Hilversum, The Netherlands), followed by 3D reconstruction and projection using Imaris 3.0 (Bitplane, Zurich, Switzerland).
Statistical Analysis
Data are presented as mean ± SEM. Significance of differences was analyzed by Student's t-test using STATA v.10.0 (StataCorp LP, College Station, TX). A value of p < 0.05 was considered significant.
Results
PTx Reversibly Disrupts Endothelial Cell Confluence
PTx treatment produced cell refolding, with cytoplasmic contraction, disruption of intercellular junctions, and loss of confluence in the cell cultures. We observed the formation of numerous intercellular gaps, leading to a reduction in the culture area covered by cells; cell-free areas were visible from 30 min post-PTx treatment (Fig. 1A). In confluent cell culture conditions, HUVEC adhered strongly to the plastic substrate and to adjacent cells, and cultures showed a cobblestone appearance (Fig. 1B). A bright field microscopy time-lapse movie showing 8 h of a confluent HUVEC culture treated with 0.1 μg/ml PTx can be downloaded from http://www.imas12.es/doc/PTx-HUVEC.avi
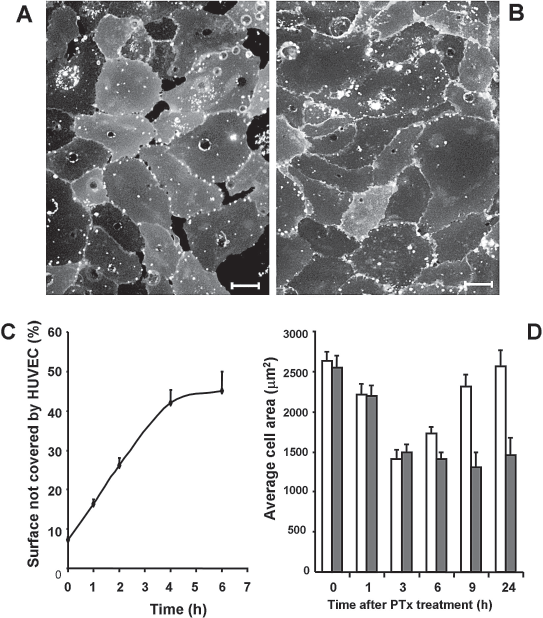
Bordetella pertussis toxin (PTx) treatment induces reversible loss of confluence in endothelial cell cultures. (A) Administration of PTx (1 μg/ml) to a confluent human umbilical vein endothelial cell (HUVEC) culture produced appearance of acellular gaps within 30 min posttreatment; (B) an untreated control monolayer remained confluent. (C) In initially confluent HUVECs, the permeability effect in the generation of acellular gaps (percentage of cell-free tissue culture surface) after PTx treatment reached maximal levels at 4 h postadministration. (D) In cells treated with PTx for 1 h, the effect was reversed by 24 h posttreatment (white bars), but not when PTx remained in culture (gray bars). Comparison of mean cell areas for these two groups shows that reversibility can be observed in the former group at 9 h posttreatment (p < 0.001 at 9 and 24 h). Scale bars: 25 μm.
When PTx was present continuously in culture, the acellular surface (Fig. 1C) and the decrease in the mean area covered by a single cell (Fig. 1D, gray bars) were maximal at 3–4 h posttreatment and persisted for 24 h without affecting cell viability (98% as measured by trypan blue staining, not shown). PTx treatment for 1 h followed by washing of cells led to a decline in the mean area covered by a single cell, which was maximal at approximately 3–4 h, began to recover after 6 h, and returned to original values at 24 h (Fig. 1D, white bars).
Cell Refolding Is Due to Reorganization of the Actin Cytoskeleton
PTx treatment of confluent HUVEC cultures induces cytoplasmic refolding, with reorganization of actin filaments in the cytoskeleton (18). In treated cells, stress fibers disappeared and actin filaments concentrated around the plasma membrane (Fig. 2A), whereas they were organized as parallel stress fibers in untreated cells (Fig. 2B).
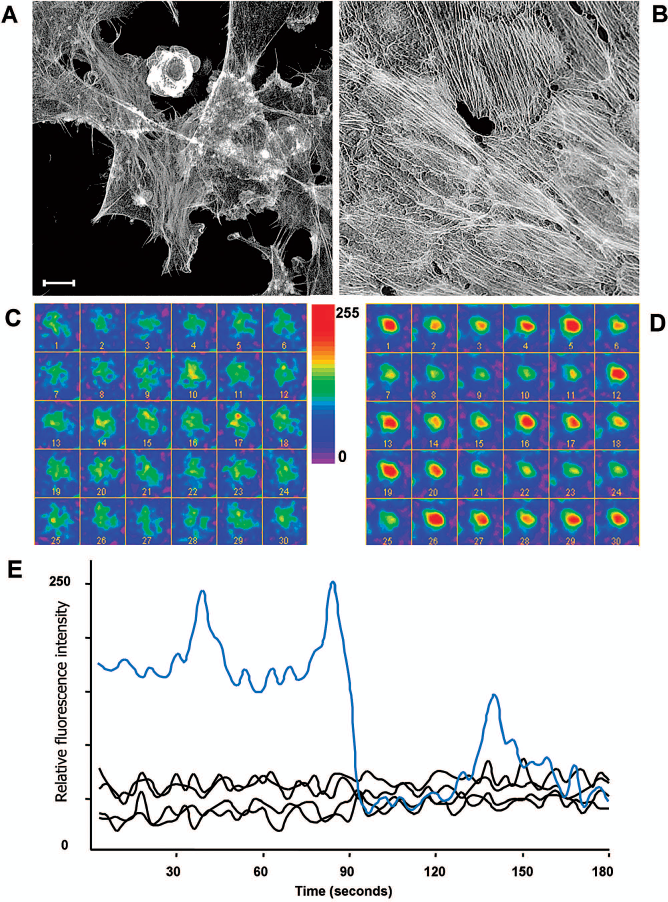
PTx-mediated induction of acellular gaps in confluent cultures involves cytoskeletal reorganization. (A, B) Projection of 12 stacked confocal images of HUVECs and Alexa-488 phalloidin stained to visualize the actin cytoskeleton: (A) HUVECs incubated 90 min with PTx, (B) control HUVEC culture. Control cells show actin filaments organized on stress fibers, well defined and parallel to fibers in adjacent cells. Stress fibers in PTx-treated cells are blurred and located beneath the plasma membrane, and cells are partially detached from the substrate. Scale bar: 15 μm. (C) Series of 30 images (acquired every 3 s) of Calcium Green-1-loaded, PTx-treated HUVECs immediately after treatment with 5 nM chemokine (C-C motif) ligand 2 (CCL2). Calcium levels were similar throughout the series. (D) In untreated control cells, calcium levels varied in response to CCL2. Right, scale of relative calcium levels (red, highest levels; dark blue, lowest). (E) Variation in Calcium Green-1 fluorescence in PTx-treated cells, representative of four experiments (black) and untreated control (blue).
PTx Treatment of Endothelial Cells Blocks Signaling Through Gi Proteins
To show that PTx blocks Gi proteins in endothelial cells, we studied signaling through CCR2, which is expressed in endothelial cells (52), is Gi protein-coupled (41). Signaling through CCR2 includes calcium mobilization, which is inhibited by PTx treatment (45). We studied intracellular calcium fluctuation by quantifying green fluorescence on Calcium Green-1-loaded HUVECs by time-lapse fluorescence microscopy. In a series of 30 images captured every 3 s beginning at 10 s post-CCL2 stimulation of a PTx-treated endothelial cell, calcium levels did not vary and were similar throughout the series (Fig. 2C); in untreated cells, we observed calcium variations in response to CCL2 (Fig. 2D). In four additional experiments, calcium fluctuations were graphed for PTx-treated HUVECs and untreated control cells in response to CCL2 (Fig. 2E).
In Vitro PTx Pretreatment of Confluent Endothelium Facilitates ES Cell Adhesion
In an in vitro cell transplant simulation system, loss of continuity in a confluent culture of PTx-treated endothelial cells creates an acellular surface that permits adhesion of other cell types (4). When seeded onto a culture of HUVECs pretreated with PTx for 90 min, ES cells adhered rapidly to HUVEC-free surfaces, and remained attached 1 h after seeding, following medium removal and washing (Fig. 3A). When seeded onto a confluent HUVEC culture (which simulates a blood vessel wall), mouse ES cells did not adhere and could be removed from culture 1 h after seeding (Fig. 3B). Adhesion to the cell-free surface was rapid; by 15 min, 50% of seeded cells had adhered to the surface (Fig. 3C).
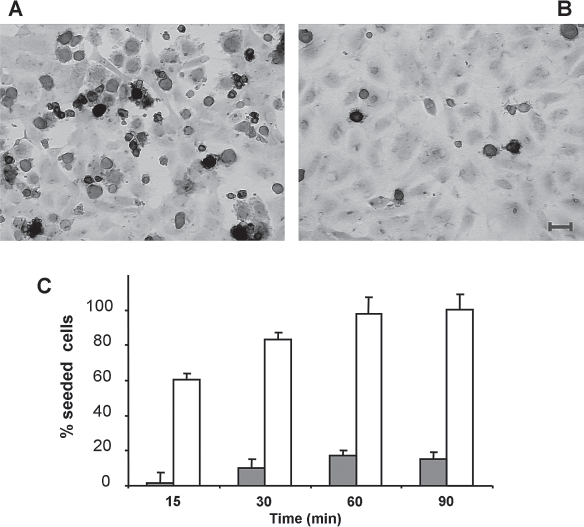
PTx treatment of confluent endothelial cell monolayers allows embryonic stem (ES) cell attachment. (A) PTx treatment (90 min) of a confluent HUVEC culture (eosin stained) induces generation of gaps that allow embryonic stem (ES) cell (blue) attachment to the substrate. (B) Untreated confluent HUVECs show no acellular gaps and do not allow ES cell attachment. (C) ES cell attachment is rapid (15 min after seeding). The percentage of attached cells (as determined by AP activity) over time is larger in PTx-treated cultures (white bars) than in controls (gray bars). Scale bar: 50 μm.
PTx Induces Loss of Confluence in Liver Endothelial Cells Both In Vitro and In Vivo
Following PTx treatment, liver endothelial cells showed refolding that left cell-free substrate areas, similar to HUVECs (Fig. 4A). Endothelial cells derived from murine liver sinusoids and cultured to confluence had a cobblestone appearance, similar to that of HUVECs (Fig. 4B). PTx also acted on the hepatic vascular tree endothelium in vivo. At 2 h after slow intraportal perfusion of PTx, FITC-G. simplicifolia lectin staining of the vascular tree showed cytoplasmic refolding similar to that observed in cell cultures; the appearance of the endothelium was discontinuous and blurred (Fig. 4C, E) whereas hepatic vessels of PBS-perfused mice showed a continuous endothelium (Fig. 4D, F).

PTx-induced endothelial reorganization in liver endothelium in vitro and in vivo. (A) PTx-treated confluent cultures of mouse liver endothelial cells, like HUVECs, showed acellular surfaces and FITC-G. simplicifolia staining, compared with (B) untreated control mouse liver endothelial cells, which showed the typical cobblestone-like endothelial appearance. (C, E) At 2 h after intraportal PTx treatment, mouse liver endothelium appears discontinuous, as visualized by FITC-G. simplicifolia staining in thick (50 μm) liver sections. (D, F) Similar sections of normal mouse liver endothelium, showing the outline of the vessels. Scale bars: 100 μm (B, D); 50 μm (F).
PTx Pretreatment of Mouse Liver Endothelium Promotes Establishment of Transplanted ES Cells
We transplanted ES cells by intraportal perfusion into livers of mice treated intrasplenically 2 h pretransplant with PBS or with PTx. Mice were sacrificed after 2 h and the degree of ES cell implantation in parenchyma was evaluated by detection of ES cell AP activity. PTx-pretreated mice showed strong ES cell AP activity in larger areas of liver sections (Fig. 5A) than did control mice (Fig. 5B). In 100-μm tissue sections, we observed a cell implantation rate of 6.36 ± 1.74% in PTx-treated mice and 1.09 ± 0.73% in untreated controls (p < 0.001) (Fig. 5C).

PTx promotes liver cell engraftment in a model of intraportal ES cell transplant. (A) After intrasplenic administration of PTx (5 μg/kg) 2 h before intraportal cell perfusion, ES cells show alkaline phosphatase (AP) activity (purple) in liver parenchyma near portal branches. (B) Fewer implanted ES cells were found in untreated controls. (C) ES cell transplant in 12 PTx-treated and 12 control mice. PTx administration prior to cell perfusion increased engraftment relative to controls (p < 0.001). (D) Phase contrast image of a 5-μm liver section from a PTx-pretreated mouse, showing individual transplanted cells (NBT positive, purple) in the periphery of small vessels. Scale bars: 100 μm (B); 25 μm (D).
ES cells transplanted in the liver were restricted to the periphery of the lobule nearest the portal vein branches, with some ES cells within the sinusoids, but not in centrilobular veins or hepatic vein branches. In mice evaluated 24 h posttransplant, implanted cells remained within the liver (Fig. 5D). Differences in AP-stained ES cells between control and PTx-treated mice were similar to those observed at 2 h (not shown), although transplanted ES cells were perivascular at 2 h and were found within the lobule at 24 h posttransplant (Fig. 5D).
Long-Term Cell Engraftment
Transplanted cells can be detected in liver parenchyma after 30 days by visualization of AP using NBT/BCIP as substrate. Although endogenous AP expression is lost in differentiating ES cells, hPAP is fused to Acer-3 under the control of the murine promoter, leading to generalized expression, particularly in liver (37). Sub-cellular Acer3-hPAP expression nonetheless differs from AP expression in cultured ES cells, as AP activity is found in Golgi and at one cell pole in transplanted ES cells. A figure showing Acer3-hPAP expression can be obtained at http://www.imas12.es/doc/Alfaro-Sup1.pdf
At 30 days posttransplant, cells in PTx-treated mice showed generalized distribution throughout the liver parenchyma (Fig. 6A), which was evident by 24 h and at 7 days after transplant (not shown). Transplanted cells in untreated controls were less numerous and were observed mainly around large vessels, with few cells in the parenchyma (Fig. 6B); in controls, distribution at 30 days was similar to that observed at 2 and 24 h and 7 days posttransplant (not shown).
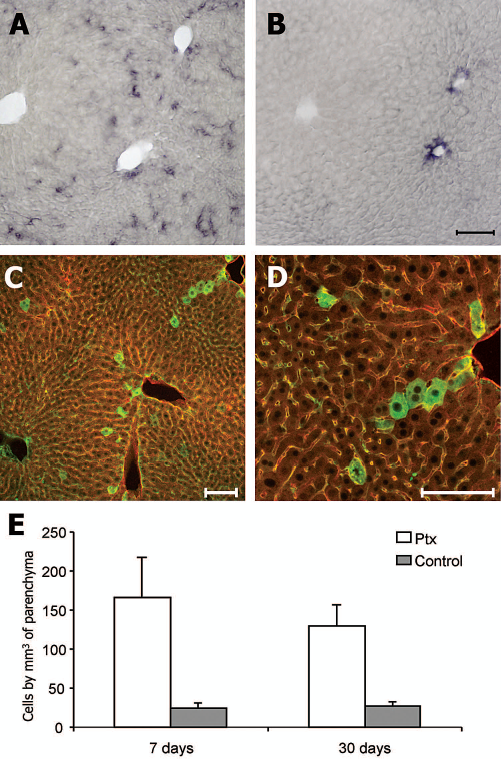
Long-term cell engraftment after ES cell transplant. (A) Cells perfused into PTx-treated mice visualized 30 days posttransplant in liver parenchyma are distributed throughout the lobule, as determined by detection of AP activity using NBT/BCIP as substrate (purple). (B) At 30 days posttransplant, cells transplanted into untreated control mice are found around the large vessels, with a few cells dispersed in parenchyma. (C) The distribution of transplanted cells in PTx-treated mice can be visualized by confocal microscopy using anti-human placental AP (hPAP; green). Lobule structure is visualized by autofluorescence, and staining of vessels with anti-CD105 (red). (D) Detail of transplanted cells in (C) at higher magnification (20x). (E) The number of transplanted cells/mm3 of liver parenchyma evaluated by image analysis in NBT/BCIP-stained sections at days 7 and 30 posttransplant in PTx-treated mice (white bars) is significantly higher than in controls (gray bars) (p < 0.05). Scale bars: 100 μm.
Transplanted cells in PTx-treated mice were also visualized by confocal microscopy using monoclonal anti-hPAP followed by a Cy2-labeled secondary antibody. Lobule structure was detected by vessel staining with antibody to the endothelial cell marker CD105 (21) followed by a Cy3-secondary antibody, and by autofluorescence (Fig. 6C, D). AP retained in cytoplasm can be visualized with anti-hPAP, but is enzymatically inactive (36,53), whereas histochemical analysis identifies only active AP, explaining the difference in subcellular distribution patterns. The number of transplanted cells in recipient liver/mm3 of parenchyma is significantly higher (p < 0.05) in PTx-pretreated mice at 7 days (166 ± 52 cells/mm3) and 30 days (129 ± 28) after transplant compared to controls (7 days, 25 ± 7; 30 days, 27 ± 4) (Fig. 6E). These differences between treated and untreated mice were very similar to those observed at 2 h posttransplant.
Discussion
To be integrated into the parenchyma, ES cells transplanted by intraportal catheter must cross the endothelial barrier, the main obstacle to their implantation (34). Here we show that PTx pretreatment of the portal vascular tree enhances early liver implant of intraportally perfused ES cells. PTx-treated endothelial cells undergo cytoplasmic folding and intercellular junctions are destabilized; vascular permeability increases due to Gi blockage-mediated activation of p38 and p42/p44 mitogen-activated protein (MAP) kinases, which induce restructuring of the actin cytoskeleton (17,18).
As the liver capillary (liver sinusoid) has no basement membrane (62), PTx-induced endothelial cytoplasmic reorganization has particular impact in liver, achieving greater permeability than in endothelia with a basement membrane. Although the permeabilizing effect of PTx has been described previously (8), our data show that this increase can facilitate liver cell implantation by intravascular infusion, and suggest a role for PTx as a engraftment enhancer. The temporal dynamics of PTx allows more rapid, efficient implantation than strategies based on cytotoxic treatments such as cyclophosphamide or irradiation (22,28,34,64). The rapid effect of PTx facilitates survival of the infused cells.
PTx pretreatment of liver endothelium does not appear to affect the migratory or implantation capacity of the transplanted cells. Although PTx blockade of chemokine receptor signaling (42) inhibits cell migratory ability by 50% at 2 h posttreatment (7), the brief period (seconds) between perfusion of transplanted cells and their entry into the capillary terminals is insufficient for blockade.
Mechanisms for cell integration into the hepatic parenchyma in the classical intraportal hepatocyte transplant model have only begun to be understood. Perfused cells are not integrated immediately, but embolize and remain trapped in the liver sinusoid, causing portal hypertension and ischemia (12). This leads to the death of many transplanted cells by oxidative stress and apoptosis due to lack of adhesion (66); dead cells are eliminated by Kupffer cells and granulocytes (26). Ischemia reperfusion injury induces the release of proinflammatory chemokines and cytokines that increase vascular permeability, allowing surviving cells to translocate through the sinusoidal endothelium and to integrate into the parenchyma (31).
Although this activation of the inflammatory response in early stages of transplant promotes the integration of surviving cells, it is counterproductive in the medium and long term, as inflammatory processes can eliminate the transplanted cells (24,26,61,65). Control of immune system activity is essential in the early stages of cell transplant, since functional blockade at the outset of the response, particularly of Kupffer cells, improves survival of transplanted cells (26,65). In its original context of B. pertussis infection, PTx inhibits early inflammatory response in two ways: it blocks chemokine production by inflamed tissues and, by preventing signaling through chemokine receptors, it impedes mobilization/entry of new immune cells (42). In addition to blocking early mechanisms of destruction, PTx aids transplanted cell survival by reducing the probability of anoikis, as it permits rapid cell extravasation and implantation (23). PTx activity is thus of interest, as it improves vascular permeability and limits the early immune response to the transplanted cells.
We used ES cells as donor cells for in vivo transplant experiments, as they are easily available from culture (59), are an appropriate choice for cell therapy because of their potential to differentiate into a variety of lineages (16), have been used effectively in transplant experiments in liver (46), can differentiate into hepatocytes in vitro and in vivo (11,13), and show strong AP expression in the plasma membrane, allowing them to be distinguished from other cell types in histological sections (19). To avoid dual insult to the portal vein, PTx or control pretreatment was administered by intrasplenic injection, as the spleen vascular supply allows rapid passage from spleen sinuses to the liver.
The number of cells transplanted to each mouse was relatively small, to ensure identification of individual cells and avoid embolus formation in blood vessels, which would complicate assessment of implantation as due to the effect of PTx or to ischemia reperfusion mechanisms secondary to embolization. To facilitate their detection, we evaluated the ES cells in thick (50–100 μm) histological sections. Implantation index values from histological section areas with AP activity correspond to a much larger volume than in standard histological sections; thicker sections showed more homogeneous ES cell distribution with less variability, permitting more accurate comparison of treated and control mice.
In mice, a single PTx dose has a transient effect on vascular permeability, with recovery at 24 h (32). Although PTx is a bacterial toxin, it showed minimum toxicity in pharmacological studies of histamine sensitization when used within the activity range (10–72 μg/kg) (60). The dose used here (5 μg/kg) is well below that which induces cardiovascular effects (20 μg/kg) (32). Additional studies are needed to establish the minimum effective dose to reduce possible adverse effects, and other potentially less toxic Gi inhibitors should be tested. It should also be determined whether interference at other points in Gi protein signaling pathways yields a similar permeabilization effect with improved clinical applicability.
Our study focused on early stages of transplant, due to the importance of rapid implantation to improve survival of transplanted cells (48). We achieved a higher degree of initial cell implant; further tests will show whether this translates to more rapid functional recovery and to an increase in the liver repopulation capacity of transplanted cells, which could allow long-term function maintenance.
Conclusion
Prior to cell transplant by intraportal perfusion, PTx treatment to block the Gi protein pathway in liver endothelium promotes rapid, efficient cell implantation in liver parenchyma and blocks chemokine receptor signaling, an essential step in early activation of the immune system.
Footnotes
Acknowledgments
This work was supported by grants from the Fondo de Investigaciones Sanitarias (2006/0483) and the Fundación Médica Mutua Madrileña (2006-075 and 2011-0444) to A.S. We thank Miguel Torres for ES cell clone B8-6C3 and advice on ES cell handling, Juan Carlos González, Ana García, and Marbella Gracia for excellent technical assistance and Catherine Mark for editorial assistance and constructive criticism of the manuscript. The authors declare no conflicts of interest.