
Abstract
Hepatocyte transplantation is considered to be an alternative to orthotopic liver transplantation. Cells can be used to bridge patients waiting for a donor organ, decrease mortality in acute liver failure, and support metabolic liver diseases. The limited availability of primary human hepatocytes for such applications has led to the generation of alternative hepatocyte-like cells from various adult stem or precursor cells. The aim of this study was to generate hepatocyte-like cells from adipose-derived mesenchymal stem cells (Ad-MSCs) for clinical applications, which are available “off the shelf.” Epigenetic changes in hepatocyte-like cells were induced by 5-azacytidine, which, in combination with other supplements, leads to significantly improved metabolic and enzymatic activities compared to nontreated cells. Cells with sufficient hepatic features were generated with a four-step protocol: 5-azacytidine (step 1); epidermal growth factor (step 2); fibroblast growth factor-4, dexamethasone, insulin-transferrin-sodium-selenite, and nicotinamide (step 3); and hepatocyte growth factor, dexamethasone, insulin-transferrin-sodium-selenite, and nicotinamide (step 4). Generated differentiated cells had higher phase I (CYP1A1/2, CYP2E1, CYP2B6, CYP3A4) and phase II activities compared to the undifferentiated cells. A strong expression of CYP3A7 and a weak expression of 3A4, as well as the important detoxification markers α-fetoprotein and albumin, could also be detected at the mRNA level. Importantly, urea metabolism (basal, NH4-stimulated, NH4- and ornithine-stimulated) was comparable to freshly isolated human hepatocytes, and unlike cryopreserved human hepatocytes, this activity was maintained after 6 months of cryopreservation. These findings suggest that these cells may be suitable for clinical application, especially for treatment of urea cycle disorders.
Keywords
Introduction
Dysfunction of the liver, which leads to hyperammonemia, can be caused by various inherited or acquired hepatic disorders; the most frequent are urea cycle disorders and advanced liver fibrosis or cirrhosis (31). In the organism, ammonia is generated by amino acid catabolism or when proteins are broken down by intestinal urease-positive bacteria. It is mostly present as ammonium (NH4+) at a physiological pH value in the blood circulation (11). Detoxification in the liver occurs mainly through conversion of NH4+ into urea via the urea cycle. Urea is nontoxic and water-soluble and can be excreted via the kidneys. In some individuals, the cycle can be interrupted by deficiencies in any of the six enzymes or two transporters involved in the urea synthesis (9). Disruption of the cycle leads to accumulation of ammonia, which is toxic to the central nervous system (10). Consequences could include lethargy, coma, and damage of the developing central nervous system with symptoms such as cognitive impairment, seizures, and cerebral palsy (20). According to estimation, these mutations have an overall prevalence of 1:8,200 in pediatric patients in the US (22). Hepatocyte transplantations have already been shown to provide a partial correction of urea cycle defects. Patients suffering from enzyme or transporter deficiencies in the urea cycle showed clinical improvement, reduced ammonia levels, and increased production of urea after hepatocyte transplantation (47, 51). Due to the limited supply of fresh primary human hepatocytes (hHeps), the best method for long-term storage of hepatocytes is cryopreservation, providing a sustained and sufficient cell supply for clinical applications (38). Unfortunately, cryopreserved human hepatocytes are easily damaged during the freezing and thawing procedure, with a significant alteration both at the morphological and functional levels (45). These losses are caused by the isolation procedure itself, initiated by oxidative stress, and the subsequent detachment procedure from the extracellular matrix, which has also been shown to promote cellular damage and apoptosis (43).
Consequently, alternative solutions are being examined in the field of hepatic cell therapy. Among these, the so-called “regenerative medicine,” based on the therapeutic potential of stem cells, is of particular interest (28, 37). Stem cells are immature cells that are able to renew themselves through mitotic division and proliferate and differentiate into different cell types. These cells can be classified into two diverse types, namely, embryonic stem cells (ESCs) and adult stem cells (ASCs). ESCs can be differentiated directly into functional hepatocytes and consequently show a tremendous potential for this application, but their clinical usage is restricted because of ethical concerns and side effects such as teratoma formation, tumor genesis, and immunogenicity (7). Induced pluripotent stem cells (iPSCs) are adult cells that are genetically reprogrammed by introducing pluripotency-associated transcription factors of somatic cells (39). Regarding hepatocytes, Huang et al. demonstrated the transduction of mouse tail tip fibroblasts with the factors Gata4, hepatocyte nuclear factor 1α (HNF1α), and forkhead box a3 (Foxa3), as well as the inactivation of p19 (24). However, the generation of transfected cells requires viral introducing procedures with frequently inefficient transduction. This method might lead to nonspecific insertion patterns that could potentially result in uncontrolled cellular proliferation, oncogenesis, or abnormal development (27).
In contrast to ESCs, adult stem cells reside not only in the early embryo but also after birth in the whole organism. During life, adult stem cells multiply by cell division to replenish dying cells and regenerate damaged tissues. Adult stem cells can be found in bone marrow, skin, adipose tissue, blood, brain, liver, or pancreas. They can generate all cell types of the organ from which they originate and also have the ability to differentiate into cell types from different germ layers (30). One type of adult stem cells is mesenchymal stem cells (MSCs), which can be derived from different organs and tissues (16). In the present study, adipose-derived mesenchymal stem cells (Ad-MSCs) were chosen as the progenitors of hepatocyte-like cells because of their multipotency, good accessibility, and scale-up properties (25). These cells are of mesoderm origin; therefore, their differentiation abilities include the adipogenic, chondrogenic, and osteogenic lineages. Interestingly, Ad-MSCs also have the ability to differentiate into nonmesoderm lineages, including the myogenic lineage leading to skeletal muscle, smooth muscle cells, and cardiomyocytes; endothelial cells; neuronal-like cells; epithelial cells; pancreatic cells; and cells with hepatic features (4, 34). The epigenetic modification of the cells is an important aspect for the hepatic differentiation of Ad-MSCs. In this study, the epigenetic changes of the cells are initiated by 5-azacytidine (AZA), known as a DNA methyltransferase inhibitor (DNMTi) (21). This cytidine analog is added during the replication and transcription into the nucleic acid strand. This integration inhibits methyltransferase and leads to demethylation of the sequence. The use of DNMTis results in cell cycle arrest, induction of apoptosis, and differentiation (15). Furthermore, the use of DNMTis lead to significantly improved phase I/II enzyme activities, higher expression of gap junctional intercellular communication markers and liver-enriched transcription factors in different cell lines, such as HeLa cells, human hepatoma cell lines, or mouse hepatocytes, as recently published by Snykers et al. (44).
Materials and Methods
Materials
The cryopreserved hHeps were provided by the Center for Liver Cell Research, Ludwig Maximilians University, after approval had been received from the Human Tissue and Cell Research Foundation, Regensburg, Germany (www.htcr.org). Dulbecco's modified Eagle's medium (DMEM-high glucose, 4.5 g/L), fetal calf serum (FCS Gold), penicillin and streptomycin, l-glutamine, HEPES, sodium pyruvate, amino acids, trypsin, and phosphate-buffered saline (PBS) were obtained from PAA Laboratories GmbH (Pasching, Austria). Human recombinant growth factors, fibroblast growth factor-4 (FGF-4), epidermal growth factor (EGF), and hepatocyte growth factor (HGF) were purchased from PeproTech (London, UK). Collagenase type II was obtained from Biochrom (Berlin, Germany). Collagenase P from Clostridium histolyticum was purchased from Roche (Mannheim, Germany). All other chemicals were obtained from Sigma (Munich, Germany).
Generation of Ad-MSCs
Ad-MSCs were isolated according to the ethical guidelines of “Klinikum rechts der Isar,” Munich, Germany. Tissues were derived from the abdomen or hip of 22 different donors (15 male and 7 female; average age of 64). Adipose tissue was cut into small pieces and washed three times with 40 ml of PBS. The tissue was digested with 0.075% (w/v) collagenase type II in PBS at 37°C for 30 min. The digestion was terminated using DMEM (2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10% FCS). The cells were then centrifuged at 450 × g for 10 min. The supernatant was aspirated, and the cells were resuspended in PBS and filtrated through a 40-μm cell strainer. The cells were seeded in 175-cm2 plastic flasks, and the medium was first changed after 24 h. After this time, the medium was changed every 3–4 days. A confluence of 80–90% complies with 1.2–1.5 million cells per 175-cm2 flask. As soon as this criterion was met, the cells were passaged for further expansion.
Isolation of Primary Human Hepatocytes
Primary hHeps were isolated from liver resections of patients with primary or secondary liver tumors with their informed consent, according to the ethical guidelines of “Klinikum rechts der Isar,” Munich, Germany. A two-step collagenase P perfusion technique was used to isolate the hHeps (36). Viability (trypan blue exclusion) of the cells was consistently above 80%. Isolated hepatocytes were cultured in Williams' medium E (10% FCS, 1 mM insulin, 15 mM HEPES, 0.8 μg/ml hydrocortisone, 100 U/ml penicillin and 100 μg/ml streptomycin, 1% l-glutamine, 1% nonessential amino acids, 1 mM sodium pyruvate) on collagen-coated culture plates with a density of 1.5 × 105 cells/cm2. Experiments were performed 1 day after the isolation.
Fluorescence-Activated Cell Sorting (FACS) Analysis
Ad-MSCs were harvested in passages 0, 1, 2, and 3 on 175-cm2 culture flasks. When the cells reached 80–90% confluence, they were trypsinized, centrifuged at 600 × g for 10 min at room temperature, and resuspended in PBS. The cells were labeled with the following monoclonal antibodies for flow cytometric analysis: mouse anti-human cluster of differentiation 14 (CD14): fluorescein isothiocyanate (FITC); mouse anti-human CD45: FITC; mouse anti-human CD73: phycoerythrin (PE); mouse anti-human HLA-DR: FITC [all purchased from Biozol (Eching, Germany)]; mouse anti-human CD105: PE from Biotech (Birmingham, AL, USA); and mouse anti-human CD90: allophycocyanin (APC) from BioLegend (San Diego, CA, USA). Acquisition and analysis were performed on a FACSCanto II from BD Biosciences (San Jose, CA, USA). All antibodies were used at a concentration of 0.5 μg/106 cells in a volume of 100 μl. An isotype control for each antibody was included in all experiments. Hematopoietic cells isolated from the blood of volunteers using the ethical guidelines of “Klinikum rechts der Isar,” Munich, Germany, served as a positive control for CD14, CD45, and HLA-DR. The software FlowJo from Tree Star, Inc. (Ashland, OR, USA) was used for data analysis.
MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide) Assay
Ad-MSCs were harvested by trypsinization, resuspended in DMEM supplemented with 10% FCS, and plated (2 × 1 0 3 cells/100 μl) in 96-well tissue culture plates. After 24 h, the cells were treated with 5, 10, or 20 μM 5-azacytidine for 24 h. After 24, 48, and 72 h, medium was replaced with 100 μl of MTT solution (0.5 mg MTT/ml) followed by incubation for 2 h at 37°C, 5% CO2. During this time, the viable cells reduced the yellow MTT to dark purple formazan crystals, which were dissolved in 100 μl of an MTT solubilization solution [10% sodium dodecyl sulfate (SDS) and 0.6% acetic acid in dimethyl sulfoxide (DMSO)]. The absorbance was measured at 570 and 690 nm.
Genomic DNA Methylation Assay
For the determination of global DNA methylation, the quantitative TaqMan-based real-time PCR system was used as previously described (52). Based on preliminary experiments in our lab, isolated Ad-MSCs were stimulated with 5, 10, or 20 μM AZA for 24 h. DNA from stimulated and untreated cells was extracted with PeqGOLD Tissue DNA Mini Kit purchased from PeqLab (Erlangen, Germany). Samples of DNA were then bisulfite-converted with EpiTect Bisulfite Kit from Qiagen GmbH (Hilden, Germany). Expression of ALU1, what is part of short interspersed elements, and long interspersed nucleotide elements (LINE1) was determined using the TaqMan Universal PCR Master Mix, No AmpErase UNG from Applied Biosystems, Roche (New Jersey, USA). For each real-time amplification, the template was equivalent to 10 ng of bisulfite-converted DNA. Measurements were performed in triplicate; the controls used were from the EpiTect PCR Control DNA Set from Qiagen. The primers (Table 1) were optimized for ABI Prism 7700 Sequence Detector Applied Biosystems, Inc. (Foster City, CA, USA). Quantification of LINE1 was normalized to the ALU1 gene within the log-linear phase of the amplification curve obtained for each probe/primer set using the ΔΔCT method.
Sequences of the Primers and Their PCR Conditions

Sequences according to Weisenberger et al. (52); primers are specific for DNA methylated sequences.
Sequence recognizes also pseudo/noncoding gene CYP2D7 in common chimpanzee.
In Vitro Differentiation of Ad-MSCs
Ad-MSCs in passage 2 were trypsinized and plated at a density of 1.5 × 104 cells/cm2 on collagen-coated dishes and cultured with DMEM (2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 1% FCS) at 37°C, 5% CO2. Differentiation of the cells was induced by sequential addition of supplements: AZA (20 μM) for 24 h; followed by EGF (20 nM) for 3 days; followed by FGF-4 (3 ng/ml), dexamethasone (DEX, 1 μM), nicotinamide (NIC, 5 mM), and insulin-transferrin-sodium-selenite (ITS, 1%) for 7 days; followed by HGF (20 ng/ml), DEX (1 μM), NIC (5 mM), and ITS (1%) for another 7 days. Ad-MSCs cultured in DMEM (2 mM -glutamine, 100 U/ ml penicillin, 100 μg/ml streptomycin, 1% FCS) at 37°C and 5% CO2 were used for comparison.
Actin Staining
To determine the morphological changes of the cells before and after differentiation, the cells were fixed (with 3.7% formaldehyde in PBS), permeabilized (with 0.2% Triton X-100 in PBS for 5 min at RT), and stained with phalloidin (1 μg/ml in PBS) and Hoechst (0.5 μg/ml in PBS) for 40 min at 37°C. After washing twice with PBS, pictures were taken with an epifluorescence microscope Eclipse TE2000-5 from Nikon (Nikon, Germany) set at 550-nm excitation wavelength and 580-nm emission wavelength for fluorescence. Hoechst staining was visualized under UV light.
RT-PCR
Total RNA was extracted using Trizol reagent according to the manufacturer's recommendations (PeqLab, Erlangen, Germany). The amount and purity of RNA were estimated by photometry. RNA integrity was examined by agarose gel electrophoresis. RNA was transcribed to first-strand cDNA using the First-Strand cDNA Synthesis Kit (Fermentas, St. Leon-Rot, Germany). The sequences of the forward and reverse primers and the conditions for RT-PCR are listed in Table 1. PCRs were performed with the melting of the cDNA at 95°C for 5 min, followed by 35 cycles at 95°C for 40 s, followed by annealing temperature for 40 s, 72°C for 40 s, and ended with a heating step at 72°C for 10 min. PCR amplicons were visualized by applying ethidium bromide in a 2% (w/v) agarose gel. cDNA from peripheral blood mononuclear cells (PBMCs) isolated from the blood of volunteers, bone marrow stem cells (BMSCs) isolated from bone marrow provided by the “Klinikum rechts der Isar” or hHeps served as positive control, and diethylpyrocarbonate (DEPC)-H2O was used as negative control. For quantification, signals were analyzed with the software ImageJ 1.42q (National Institute of Health, Maryland, USA).
Freezing and Thawing of Ad-MSCs, Hepatocyte-Like Cells, and hHeps
With a method frequently used in our group, both differentiated and undifferentiated Ad-MSCs were cryopreserved for 6 months. The cells were trypsinized and centrifuged at 420 × g, 4°C for 10 min. The cell pellet was resuspended in DMEM (40%), FCS Gold (50%), and DMSO (10%) to a final concentration of 1 × 106 cells/ml. The cells were immediately frozen at −20°C for 1 day, followed by −80°C for 2 days, and then transferred into a liquid nitrogen tank for long-term storage. For thawing, the vials were removed from the liquid nitrogen storage tank, and the cells thawed rapidly by immersing the vials in a 37°C water bath. Immediately after thawing, DMSO was removed by successive dilution in preheated medium, followed by centrifugation at 600 × g for 10 min. The supernatant was aspirated, and the cells were resuspended in medium. Medium was changed after 24 h.
Culture and Plating Efficiency
Thawed cells were plated in collagen-coated 24-well plates at a density of 1.5 × 105 cells/well in 1 ml of medium. The first medium change was 24 h after plating. After this time, nonadherent cells were counted, and the protein content of adherent and nonadherent cells was determined. The extent of attachment was calculated as a percentage of cellular protein remaining compared to the amount initially plated.
Cytochrome P450 Activity Assays
Modified fluorescence-based cytochrome P450 (CYP) assays were performed by incubating intact cells with selected substrates for 120 min at 37°C (19). All conditions and substrates used are listed in Table 2. The amount of product formed was normalized to the total protein content by sulforhodamine B (SRB) staining measured at 565 and 690 nm. Absorbance and fluorescence were measured using a FLUOstar Omega purchased from BMG Labtech (Offenburg, Germany).
Reaction Condition for P450 Isozyme Measurement

7-EC, 7-ethoxycoumarin; 7-HC, 7-hydroxycoumarin; AMMC, 3-(2-N, N-diethylaminoethyl)-7-hydroxy-4-methylcoumarin; EFC, 7-ethoxy4(trifluoromethyl) coumarin; HFC, 7-hydroxy-4(trifluoromethyl)coumarin; MFC, 7-methoxy-4(trifluoromethyl)coumarin; BFC, 7-benzyloxy-4(trifluoromethyl) coumarin; CHC, 3-cyano-7-hydroxycoumarin; AHMC, 3-[2-(diethylamino)ethyl]-7-hydroxy-4-methylcoumarin; MCB, monochlorobimane; 4-MU, 4-methylumbelliferone.
Urea Measurement
The cells were washed with PBS and then incubated for 24 h with PBS containing 1 mM MgCl2 and 1 mM Na-pyruvate in the presence or absence of 0.3 M NH4Cl or 0.3 M NH4Cl and 0.1 M ornithine. After this time, 80 μl of the supernatant was incubated with 60 μl of O-phthaldehyde solution (1.5 mM O-phthalaldehyde, 4 mM Brij-35, 0.75 M H2SO4) and 60 μl of NED reagent [2.3 mM N-(1-naphthyl)ethylenediamine dihydrochloride, 0.08 M boric acid, 4 mM Brij-35, 2.25 M H2SO4] for 2 h at 37°C (26). Absorbance was measured at 505 nm and compared to the dilution series of the urea stock solution (100 μg/ml).
Glucose Measurement
Cells were washed twice with PBS before incubating for 24 h with PBS (1 mM MgCl2, 1 mM Na-pyruvate in the presence or absence of 10 mM Na-l-lactate). A volume of 100 μl of the supernatant was incubated with 150 μl of GLOX solution (250 mM Tris, 0.2 mM EDTA, 0.04% glucose-oxidase, 0.007% peroxidase, 0.01% O-dianisidine, pH 8.0) for 2 h at 37°C. The absorbance was measured at 420 nm and compared to the dilution series of the glucose stock solution (600 μmol).
Statistics
Results are given as mean ± SEM. One-way analysis of variance (ANOVA) with Bonferroni's multiple comparison test and Student's unpaired two-tailed t test were performed using GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, CA, USA). The value of p < 0.05 was taken as the minimum level of significance.
Results
Characterization of Ad-MSCs
Ad-MSCs Express the Required CD Marker Pattern in Passage 3
Mesenchymal cells isolated from human adipose tissue displayed typical features of MSCs, such as plastic adherence, proliferative behavior, and a characteristic spindle shape. The population doubling time (PDT) of the cells increases over the passages from 1.48 to 4.73 days in passage 7 (data not shown). In passage 3, the cell population was homogenous, and the expression of CD13, CD29, CD44, and CD166 was detected at mRNA level (Fig. 1A). The adhesion molecules CD31 and CD34 and the marker CD147, important for spermatogenesis, embryo implantation, neural network formation, and tumor progression, were undetectable in Ad-MSCs of passage 3.
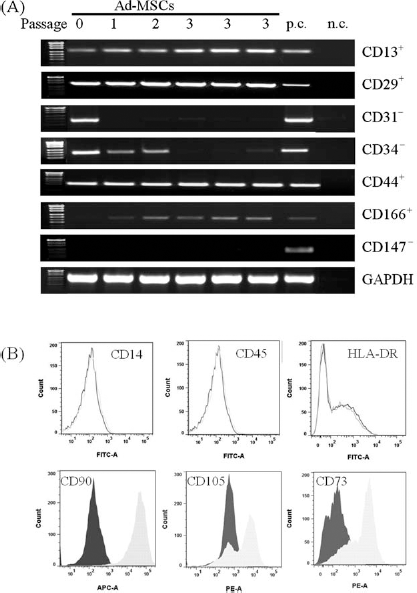
Adipose-derived mesenchymal stem cells (Ad-MSCs) express required cluster of differentiation (CD) markers. (A) Phenotype of Ad-MSCs from passages 1 to 3 according to RT-PCR and fluorescence-activated cell sorting (FACS) analysis. (B) Cells incubated with the respective fluorescein isothiocyanate (FITC)-, allophycocyanin (APC)-, or phycoerythrin (PE)-labeled antibodies and analyzed by flow cytometry. Light gray shows curve-specific antibody; dark gray shows curve isotype control. Representative analyses from three different experiments are shown (N = 3, n = 4). hHeps, human hepatocytes; p.c., peripheral blood mononuclear cells (PBMCs) or bone marrow stem cells (BMSCs) served as positive control; n.c., DEPC-H2O served as negative control.
CD markers 45, 73, 90, and 105, as well as HLA-DR, were analyzed by flow cytometry to confirm the specific pattern of MSCs (Fig. 1B). FACS analysis revealed that the cell population in passage 3 was positive for the markers CD73, CD90, and CD105 (92.63%, 84.49%, and 86.17%, respectively) and negative for CD14, CD45, and HLA-DR (1.28%, 1.88%, and 1.06%, respectively). The use of AZA did not change these findings (data not shown).
AZA Reduces Global Methylation in Ad-MSCs
Preliminary data of our group showed no toxicity when incubating with 5, 10, or 20 μM AZA over 72 h (data not shown). For an investigation of the global methylation status, isolated Ad-MSCs were stimulated with 5, 10, or 20 μM AZA for 24 h (Fig. 2A). Quantification of the global methylation status was realized by normalizing the LINE1 values to the ALU1 values. The global DNA methylation status was significantly reduced by up to 24.6 ± 12.5% with 5 μM AZA, by up to 26.5 ± 17.2% with 10 μM AZA, and by up to 28.7 ± 7.2% with 20 μM AZA in comparison to untreated cells. In view of these results, Ad-MSCs were pretreated with 20 μM AZA for 24 h.

Global methylation status and morphological changes of Ad-MSCs. (A) Methylation status of Ad-MSCs analyzed after 24 h of incubation with different concentrations of 5-azacytidine (AZA) (N = 4, n = 6). EpiTect PCR Control DNA was used as control. (B) After hepatic differentiation, cells showed a rounder and more hexagonal shape. For comparison, human hepatocytes are shown.
Hepatic Differentiation of Ad-MSCs
AZA Improves Significantly the Hepatic Differentiation of Noncryopreserved Ad-MSCs
Following AZA exposure, cells were then incubated with a mixture of FGF-4, ITS, NIC, and DEX for additional 13 days. The hepatic differentiation process itself (i.e., the addition of growth factors, NIC, and DEX) improved significantly the metabolism of urea and glucose, as well as important phase I/II enzyme activities of the Ad-MSCs (Table 3). Hepatocyte-like cells pretreated with AZA exhibited a fivefold higher production of glucose than non-pretreated cells and reached 25% of the production in hHeps. Furthermore, urea production by hepatocyte-like cells pretreated with AZA was twofold higher than non-pretreated and was comparable to hHeps. CYP1A1/2 activity was threefold higher in AZA-pretreated than in non-pretreated cells, and the activity was the same as that in hHeps. A slightly higher enzymatic activity of CYP2E1 was detectable in the cells pretreated with AZA compared to the untreated ones. CYP2B6 activity was also significantly increased (fivefold) by pretreatment with AZA. In addition, phase II enzyme conjugation of 7-hydroxy-4-(trifluoromethyl)coumarin (HFC), 3-[2-(diethylamino) ethyl]-7-hydroxy-4-methylcoumarin (AHMC), and 3-cyano-7-hydroxycoumarin (CHC) was also improved by pretreating the cells with AZA prior to the differentiation process. HFC, AHMC, and CHC conjugation rates were increased by 5-, 16-, and 12-fold in AZA-pretreated cells than in non-pretreated cells, respectively. As a result, subsequent studies always included AZA treatment for 1 day before the differentiation protocol.
Analysis of Glucose and urea Metabolism, Phase I and Phase II Enzyme Activities of Differentiated Noncryopreserved Cells and the Effect of Pretreatment With 5-Azacytidine (AZA)

Control cells were adipose-derived mesenchymal stem cells (Ad-MSCs).
p < 0.05,
p < 0.01, and
p < 0.001 compared to cells without AZA pretreatment and human hepatocytes (hHeps) (N = 3, n = 6).
The Morphology Changes Due to the Differentiation of the Cells
After hepatic differentiation, the cells of adipose origin exhibited a compact hexagonal shape and were similar in appearance to hHeps (Fig. 2B). Adherent hHeps have a smaller size compared to the hepatocyte-like cells, but after trypsinization, the size of all cells was comparable to Ad-MSCs (14.2 ± 3.3 μm, N = 3, n = 1; data not shown).
Differentiation Improves RNA Expression of Phase I Enzymes and Hepatic Markers
Important phase I enzymes and hepatic markers were detectable at the mRNA level (Fig. 3A). Expression of CYP1A1 and CYP1A2 was clearly detectable in both Ad-MSCs and hepatocyte-like cells; however, there was a low expression of CYP2D6, CYP2B6, and CYP3A4 in these cells compared to hHeps. A strong expression of CYP3A7 was visible in undifferentiated and differentiated cells. Differentiated cells had a higher expression of CYP7A1 and α-fetoprotein than Ad-MSCs. Moreover, expression of albumin was detectable in both Ad-MSCs and hepatocyte-like cells. Quantification of the PCR signals showed that there was a 1.6-, 1.44-, and 1.37-fold higher density of CYP1A2, CYP3A7, and CYP7A1 in the hepatocyte-like cells than in undifferentiated Ad-MSCs (Fig. 3B). There were no significant differences in the densities of CYP1A1, CYP3A4, α-fetoprotein, and albumin between Ad-MSCs and hepatocyte-like cells.

Expression of Phase I enzymes and hepatic markers. (A) RNA expressions of Ad-MSCs (no. 1) and hepatocyte-like cells (no. 2). hHeps, human hepatocytes; n.c., negative control (DEPC-H2O). (¤, double the amount; ¤¤, three times the amount; ¤¤¤, four times the amount of 30 ng RNA template from Ad-MSCs and hepatocyte-like cells). Representative analyses from three different experiments are shown. (B) Densities of RNA expressions of Ad-MSCs (white bars) and hepatocyte-like cells (gray bars) are displayed. Values are expressed as a percentage of hHeps. **p < 0.01 compared to Ad-MSCs, Ad-MSCs, hepatocyte-like cells, and hHeps (N = 3; n = 1).
Cryopreservation of Ad-MSCs, Hepatocyte-Like Cells, and hHeps
Viability and Attachment Efficiency
The viability of Ad-MSCs that had been cryopreserved for 6 months was 41.3 ± 6.1%, and 58.3 ± 1.1% of the living cells attached after 24 h. The viability of hepatocyte-like cells after cryopreservation was 59.2 ± 10.3%, and 67.3 ± 18.2% of the living cells attached after 24 h. By comparison, hHeps cryopreserved for up to 6 months were 45.6 ± 3.0% viable, and 37.6 ± 3.5% of them attached after 24 h.
Differentiation of Ad-MSCs Prior to Cryopreservation Conserves Phase I Enzyme Activities
Cryopreserved Ad-MSCs that were differentiated after cryopreservation (postdifferentiated) had significantly lower CYP activities than cells that had been differentiated to hepatocyte-like cells prior to cryopreservation (predifferentiated) (Fig. 4A). CYP1A2 and CYP3A4 activities were 4- and 25-fold higher in predifferentiated hepatocyte-like cells than in postdifferentiated cells. Both activities were comparable to the enzyme activity of cryopreserved hHeps. Furthermore, CYP2B6 and CYP2A1 activities in predifferentiated hepatocyte-like cells were 3.8-fold higher than in postdifferentiated cells. Predifferentiated hepatocyte-like cells had a 3.8-fold higher activity of CYP2B6 than cryopreserved hHeps, but only 1.2-fold higher CYP2A1 activity. With respect to phase II enzyme activities, there were no significant differences between predifferentiated and postdifferentiated hepatocyte-like cells (Fig. 4B). The rates of 7-hydroxycoumarin (7-HC) and 4-methylumbelliferone (4-MU) conjugation were the same regardless of when the cells were differentiated and were a third of the activity of cryopreserved hHeps. By contrast, although the rates of HFC and resorufin conjugation were also comparable in pre-and postdifferentiated cells, the activities were 3- and 1.5fold higher than those in cryopreserved hHeps.

Phase I/II enzyme activities of differentiated cryopreserved cells. (A, B) Phase I enzyme activities of CYP1A1/2, CYP3A4, CYP2B6, and CYP2A1 and phase II enzyme activities of 7-HC, 4-MU, HFC, and Res (see Table 2 for definitions) conjugation in Ad-MSCs that had first been cryopreserved and then differentiated into hepatocyte-like cells and from predifferentiated cryopreserved hepatocyte-like cells are shown. All values are expressed as a percentage of cryopreserved hHeps. Enzyme activities were normalized to total protein contents measured by sulforhodamine B (SRB) staining. **p < 0.01 compared to cryopreserved and differentiated Ad-MSCs. Cryopreserved Ad-MSCs, hepatocyte-like cells, and hHeps (N = 3; n = 4).
Predifferentiation of the Cells Conserves Glucose Production During Cryopreservation
Glucose metabolism in the cryopreserved hepatocyte-like cells was comparable to noncryopreserved differentiated cells and was twofold higher than that in cryopreserved hHeps (Fig. 5A). Glucose production in cryopreserved hHeps was significantly lower (7.8-fold) than that in freshly isolated hHeps. This was determined according to basal and lactate-stimulated levels. Surprisingly, no glucose production could be detected in Ad-MSCs that were differentiated after 6 months of cryopreservation.

Metabolic capacities of cryopreserved Ad-MSCs and hepatocyte-like cells. (A) Glucose formation rates at basal levels (white bars) and after 24 h of stimulation with lactate (light gray bars) are shown. (B) Urea formation rates at basal levels (white bars), after 24 h of stimulation with NH4Cl (light gray bars), and after 24 h of stimulation with NH4Cl and ornithine (dark gray bars) are shown. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to hHeps. Cryopreserved Ad-MSCs, hepatocyte-like cells, and hHeps (N = 3; n = 4); unfrozen hepatocyte-like cells and hHeps (N = 9; n = 4). Cryo., 6-month cryopreservation; diff., hepatic differentiated.
Cryopreservation Has No Effect on the Urea Metabolism
Urea production was the same in pre- and postdifferentiated and noncryopreserved hepatocyte-like cells (Fig. 5B). This was determined according to basal and NH4Cl-stimulated (in the presence or absence of ornithine) levels. The production of urea in hepatocyte-like cells was equivalent to the levels measured in mature freshly isolated hHeps and was up to twofold higher than that in cryopreserved hHeps.
Discussion
Inadequate or delayed donor availability restricts the ability of orthotopic liver transplantation, and some patients may not be good candidates for this procedure because of underlying medical, technical, or psychosocial contraindications. Therefore, cell-based therapies are an attractive tool for bridging different liver defects, such as metabolic dysfunctions or acute liver failures (8). Hepatocyte transplantation, a less invasive alternative to orthotopic liver transplantation, is mainly indicated for inborn metabolic disorders, in particular, urea cycle effects and Crigler–Najjar syndrome type 1 (3, 23). Meyburg et al. describe the clinical use of cryopreserved hepatocytes for the treatment of urea cycle disorders (32). They could transplant cryopreserved hepatocytes from a 9-day-old neonate intraportally in four children suffering from urea cycle disorders. However, these cells are rarely available because the donor organs normally provided for isolation are mostly of marginal quality, and the quality of the isolated cells varies considerably. In addition, for hepatocyte transplantation, some immunosuppressive interventions are required to prevent rejection of the transplanted cells (41). As a result of these restrictions, researchers are trying to find alternatives based on stem cell technologies, such as MSCs (2, 35).
Different tissue sources can be used for MSCs, such as bone marrow, umbilical blood, placental tissue, or adipose tissue. The latter is easily accessible compared to other tissues, shows a good proliferation capacity, and can be isolated in larger quantities that yield a large amount of cells. We estimate that only 5 g of adipose tissue is needed to generate 35 million cells that can be differentiated. Of course, this amount is not sufficient for cell transplantation for which 30–100 million cells/kg body weight might be needed; however, new techniques such as bioreactors are already in test phases to overcome this limitation. For example, a 50-ml rotary bioreactor increased the cell density of BM-MSCs by ninefold after 8 days of culture, resulting in a total number of 8.93 ± 0.41 × 106 cells/ml (12).
Characterizing a cell as a mesenchymal stem cell requires minimal criteria including adherence to plastic; multipotent differentiation potential; expression of surface markers such as CD105, CD90, and CD73; and lack of expression of CD14, CD34, CD45, and HLA-DR (18). Based on this definition, the MSCs isolated from human adipose tissue yielded a cellular fraction with genuinely pure MSCs. Ad-MSCs were used in passage 3 to avoid the risk of transdifferentiation, spontaneous transformation, and increasing PDT, which occurs in later passages.
In the present study, we used a differentiation protocol that included AZA, EGF, NIC, FGF-4, DEX, ITS, and HGF. Epigenetic changes during the differentiation process are key to the success of the process. AZA, a DNA methyltransferase inhibitor, initiates the dedifferentiation by decreasing the global methylation status of the cells, and therefore, genes are more prone to transcription (40). We could detect a decrease of the global methylation status by using 20 μM AZA, which is in agreement with earlier findings (6). However, more interestingly, epigenetic changes support the hepatic differentiation and lead to an increase in metabolic and enzymatic activities in human cells. This is substantiated by the findings of different other groups working with rodent or human mesenchymal stem cells of different origin (5, 42, 46). The combination of epigenetic changes plus a distinct differentiation protocol results in cells with specific hepatic features. The detectable expression of albumin and α-fetoprotein in our cells are additional indicators for the successful hepatic differentiation. Moreover, the hepatocyte-like cells exhibit activities or the expression of important enzymes such as CYP3A4/7, CYP2D6, CYP2B6, CYP1A2, and CYP2E1, which comprise more than 90% of drug oxidation in humans (54). The markedly detectable expression of CYP3A7—a typical marker of hepatic progenitors—suggests that our hepatocyte-like cells developed the ability to metabolize relevant drugs (53). The activities of phase II drug-metabolizing enzymes, which play an important role in biotransformation of endogenous compounds and xenobiotics, are less prominent in hepatocyte-like cells than in primary hHeps but these were strongly increased by the differentiation process, including epigenetic changes. We have tried to correlate our results of the individual donors with the patients' demographic data. Nevertheless, no correlation was observed between the hepatic differentiation efficiency and the patients' age (data not shown).
During 6 months of cryopreservation, the loss of Ad-MSCs and hepatocyte-like cells ranged between 35% and 65%. A comparable loss of cell viability was also observed for the hepatocytes following prolonged cryopreservation. This is also described by Chesne et al. (14), who cryopreserved hepatocytes from various animal species and human donors and reported a decreased cell viability of up to 25% and a reduced attachment efficiency of up to 50%. Many groups tried to decrease these losses with different storage solutions or complex freezing procedures, but these have generally only resulted in viabilities between 52% and 75% and attachment efficiencies of between 40% and 48% (1, 49). In addition, glucose and urea metabolism in hHeps was significantly decreased after cryopreservation. De Loecker et al. (17) reported similar losses in these activities, whereby glycogenolysis was decreased by up to 30% and by approximately 47% 1–2 days postthaw. The decline of urea metabolism, which is a sensitive marker for cell viability since it requires a high energy demand from cells, as well as coupled transport in different organelles, was also shown by Chen et al., who cryopreserved pig hepatocytes (13). By contrast, hepatocyte-like cells conserved much of the ability to metabolize glucose and urea. Urea cycle dysfunctions lead to increased toxic ammonia levels in the body, and as already shown from hepatocyte transplantations, the defective hepatocytes may benefit from the support of hepatocyte-like cells (33).
These studies emphasize the importance of differentiation prior to cryopreservation of the cells because a significant loss of metabolic and enzymatic activities was observed when Ad-MSCs were first cryopreserved and then differentiated after thawing. The cryostability of MSCs was also observed by Tokumoto et al. (50), who could not detect any effects of cryopreservation on the attachment efficiency, proliferation, and osteogenic differentiation of BM-MSCs. Likewise, cryopreserved adipogenic-differentiated Ad-MSCs have been shown to be comparable with freshly differentiated cells with respect to the regeneration of adipose tissue (29). Our findings provide the possibility to generate cells that are always available and have hepatic features for specific clinical applications.
In conclusion, the generated hepatocyte-like cells derived from adipose tissue offer a promising alternative approach to the treatment of urgent metabolic liver dysfunctions. These cells have a number of advantages: (i) preservation of important metabolic functions and many enzymatic activities during cryopreservation (48); (ii) immediate and constant availability; and (iii) the possibility for autologous application without the necessity of lifelong immunosuppressive therapy.
Footnotes
Acknowledgments
This work was partially supported by the Federal Ministry of Research (BMBF 01GN0984) and Embryonic Stem cell-based Novel Alternative Testing Strategies (ESNATS 201619). We would sincerely like to thank the Human Tissue & Cell Research Foundation for making human tissue available for research. We would also like to thank Fritz Seidl for proofreading this paper and Marina Unger for her technical support. Funding Sources: BMBF 01GN0984 and ESNATS 201619. The authors declare no conflict of interest.