
Abstract
Exogenous cell replacement represents a potent treatment option for Parkinson's disease. However, the low survival rate of transplanted dopaminergic neurons (DA) calls for methodological improvements. Here we evaluated a method to combine transient genetic modification of neuronal progenitor cells with an optimized cell culture protocol prior to intrastriatal transplantation into 6-hydroxydopamine (6-OHDA) unilateral lesioned rats. Plasmid-based delivery of brain-derived neurotrophic factor (BDNF) increases the number of DA neurons, identified by tyrosine hydroxylase immunoreactivity (TH-ir), by 25% in vitro, compared to enhanced green fluorescence protein (EGFP)-transfected controls. However, the nucleofection itself, especially the cell detachment and reseeding procedure, decreases the TH-ir neuron number to 40% compared with nontransfected control cultures. To circumvent this drawback we established the colayer method, which contains a mix of nucleofected cells reseeded on top of an adherent sister culture in a ratio 1:3. In this setup TH-ir neuron number remains high and could be further increased by 25% after BDNF transfection. Comparison of both cell culture procedures (standard and colayer) after intrastriatal transplantation revealed a similar DA neuron survival as seen in vitro. Two weeks after grafting TH-ir neuron number was strongly reduced in animals receiving the standard EGFP-transfected cells (271 ± 62) compared to 1,723 ± 199 TH-ir neurons in the colayer group. In contrast to the in vitro results, no differences in the number of grafted TH-ir neurons were observed between BDNF, EGFP, and nontransfected colayer groups, neither 2 nor 13 weeks after transplantation. Likewise, amphetamine and apomorphine-induced rotational behavior improved similarly over time in all groups. Nevertheless, the colayer protocol provides an efficient way for neurotrophic factor release by transplanted progenitor cells and will help to study the effects of candidate factors on survival and integration of transplanted DA neurons.
Keywords
Introduction
Parkinson's disease (PD) is characterized by the progressive degeneration of dopaminergic (DA) neurons of the substantia nigra, which causes cardinal motor symptoms of bradykinesia, rigidity, and resting tremor. One therapeutic approach to substitute the lost DA neurons represents the cell replacement strategy in which fetal ventral midbrain-derived DA neurons are transplanted into the PD striatum. Both animal experiments and open-label clinical trials have revealed morphological and functional integration of grafted DA cells into the host striatum. Although, results of two double-blind placebo-controlled clinical trials were disappointing, several improvements, such as tissue handling, transplantation procedure, immunosuppression, and patient selection, will be considered for follow-up clinical trials (10,25, 26,48).
To overcome the limited supply of fetal donor tissue, embryonic stem cells (ESCs) and neural progenitor cells (NPCs), respectively, could be alternative cell sources. Because ESCs display disadvantages like chromosomal aberrations in midterm cultures (14) and tumor formations (e.g., teratoma-like structures) when grafted into adults (7,35), NPCs are of high interest to develop restoration strategies further (15,30,34,41). However, irrespective of the cell source, the major problem of the transplantation approach remains the low survival rate of 1–20% of the transplanted DA neurons (11).
Neurotrophic factors have been described with regard to their survival-promoting and/or protecting activities on DA neurons (20,24). Neurotrophic factors can be applied either to prevent further death of endogenous DA neurons or can be used in cellular replacement approaches. Although, several neurotrophic factors were successfully tested in vitro with regard to increased neurite outgrowth and/or survival of DA neurons, only some reveal physiological relevance in the nigrostriatal system (5,19,38). In addition to glial-derived neurotrophic factor (GDNF) and basic fibroblast growth factor (bFGF-2), the neurotrophin family member brain-derived neurotrophic factor (BDNF) is one of the best characterized neurotrophic factors for DA neurons in vitro and in vivo (3,23,29,31,46).
Several strategies have been applied to employ neurotrophic factors to protect endogenous DA neurons or increase survival of intrastriatal DA grafts, respectively. For example intrastriatal or mesencephalic transplanted fibroblasts, which have been genetically modified to secrete BDNF, have confirmed the neuroprotective effect of BDNF against subsequent 6-hydroxydopamine (6-OHDA) or 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) injections (16,17,27). In restorative approaches with intrastriatally transplanted DA neurons recombinant mature BDNF protein has been either applied as simple in vitro pretreatment (22,52) or administered in vivo via infusion or daily injections into the host striatum close to the transplantation site (40,50,51). In summary, these studies revealed that continuous BDNF administration for up to 28 days after transplantation increases tyrosine hydroxylase immunoreactivity (TH-ir) fiber outgrowth and functional recovery, whereas pretreatment with BDNF in vitro alone was insufficient. Interestingly, optimal effectiveness of BDNF infusion, administered in the third and fourth week after transplantation into adult rat brains, coincided with the highest levels of striatal BDNF expression seen during postnatal development at day P27, which corresponds well with the age of the transplanted cells (50).
The aim of the present study was to genetically modify neural progenitor cells to overexpress BDNF in order to increase their survival after transplantation. We developed an efficient in vitro protocol for preparation and nonviral transfection of ventral mesencephalic-derived neural progenitor cells and evaluated their survival (2-week time point) and functional and morphological integration (13-week time point) after intrastriatal transplantation using the unilateral 6-OHDA rat model. We present here a promising approach to increase graft survival and to identify transplanted cells also months after transplantation.
Materials and Methods
Expression Vectors
Plasmids were derived from pCAGGS-empty (R399) provided by Dr. Hitoshi Niwa, RIKEN Center for Developmental Biology, Japan (37), which contains the CAG promoter composed of cytomegalovirus (CMV) immediate early enhancer and chicken β-actin promoter. The enhanced green fluorescence protein (EGFP) expression plasmid pCAGGS-EGFP (R410) was a gift of Dr. James R. Whiteford, Imperial College London, UK (2). The pCAGGS-EGFP-FLAG plasmid (R412) was constructed by insertion of a 3xFLAG sequence (encoding the SRGSRAYKDHDGDYKDHDIDYKDDDDK epitope tag) at the C-terminus of the EGFP sequence. The coding sequence of BDNF (NM_012513, GenBank) was amplified by polymerase chain reaction (PCR) from rat embryonic brain cDNA using RnBDNF_F:
Western Blot
Protein samples were prepared from cultured cells by a radioimmune precipitation assay buffer [20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM β-glycerophosphate, 10 mM sodium fluoride, 2 mM ethylenediamine-tetraacetic acid (EDTA) disodium salt, 1% Triton X-100, 1% deoxycholate, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride, 1x protease inhibitor (Roche), 1x phosphatase inhibitor (Roche)]. After 5 min of boiling in Laemmli buffer the samples were separated by sodium dodecyl sulfate (SDS) polyacrylamide gel electrophoresis (PAGE) [12% gel for BDNF and 8% gel for tyrosine kinase receptor B (TrkB)] and transferred electrophoretically to nitrocellulose membranes (Hybond ECL, Amersham). The membranes were probed with anti-BDNF antibody (sc-546, Santa Cruz, 1:1,000), anti-FLAG-M2 antibody (F1804, Sigma, 1:3,000), anti-TrkB antibody (80E3, Cell Signaling, 1:1,000), anti-phosho-TrkB-Tyr706/707 (C50F3, Cell Signaling, 1:1,000). After washing, the membranes were incubated with anti-rabbit/mouse antibodies conjugated to horseradish peroxidase (Amersham Bioscience, 1:4,000), followed by a chemiluminescence reaction (Immobilon Western kit, Millipore) detected on a Chemiluminescence Imager system (Intas).
Animals and Lesion Surgery
Sprague-Dawley rats obtained from Charles River (Germany) were housed four to a cage under 14:10 h light/dark cycle with ad libitum access to food and water. To generate the hemiparkinsonian rat model, adult females weighing 220–260 g were anesthetized with chloral hydrate (370 mg/kg, IP), followed by two stereotaxic injections of 6-OHDA (3.6 μg/μl in 0.2 mg/ml l-ascorbate saline) targeting the right medial forebrain bundle. The following coordinates were used (values in millimeters with reference to bregma and dura): first 2.5 μl of 6-OHDA at AP −4.4, LAT −1.2, VERT 7.8, tooth bar −2.4 and second 3 μl of 6-OHDA at AP −4, LAT −0.8, VERT −8, tooth bar +3.4. The injection rate was 1 μl/min, and the cannula of the 10-μl Hamilton syringe was left in place for an additional 3 min before slowly retracting it. Animals were not immunosuppressed as it is not necessary for intraparenchymal allografts of fetal mesencephalic cell suspensions in this Parkinson model (9). All experimental protocols followed German law on animal care and were approved by the Bezirksregierung Hannover, Germany.
Drug-Induced Rotational Behavior
Three and 6 weeks after 6-OHDA lesion and 3, 6, 9, and 12 weeks after intrastriatal transplantation apomorphine- and amphetamine-induced rotation was monitored in automatic rotometer bowls (47). The animals were injected SC with the postsynaptic dopamine receptor agonist apomorphine (0.05 mg/kg in ascorbate saline) and right and left full body turns were monitored over 40 min. After 4–7 days of recovery time the presynaptic dopamine-releasing and reuptake inhibiting drug d-amphetamine (5 mg/kg in saline) was injected IP and turning behavior was recorded for 90 min (44). Animals displaying more than 4 turns/min after apomorphine and more than 7 turns/min after amphetamine application were included in the cell transplantation experiments. Based on their rotational score animals were divided into equal groups. All behavioral tests were performed and analyzed by observers blinded to the groups.
Cell Culture
Primary cultures of DA progenitor cells were prepared from dissociated cell suspensions of dissected ventral mesencephalons (VM) of embryonic day 12 (E12) rat embryos (6,33). A detailed description of the procedure has been reported previously (44). After determining viable cell number using the trypan blue dye exclusion assay (Sigma), cells were seeded on polyornithine (0.1 mg/ml)/laminin (6 μg/ml)-coated multi-well plates. For in vitro differentiation of DA neurons three cell culture media were used. The adhesion medium contained Dulbecco's modified Eagle's medium/Ham's F-12 medium (DMEM/F12, Gibco), 3% fetal calf serum (FCS, PAA), 20 ng/ml FGF2 (Preprotech), 1x B27 (Gibco), 1x N2 (Gibco), 1 mM sodium pyruvate, 0.25% (w/v) bovine serum albumin (BSA, Sigma), 2 mM glutamine. The proliferation medium resembles the composition of adhesion medium omitting FCS and B27 supplement. The differentiation medium contained DMEM/F12, 0.25% (w/v) BSA, 1x B27, 1% FCS, 100 μM ascorbic acid, 2 mM glutamine.
The freshly prepared cell suspensions were cultured for 1 day in adhesion medium, followed by 3 days in proliferation medium. On the fourth day in vitro (DIV4) cells were transfected (see below) and medium was switched to differentiation medium for 2–6 days. For in vitro experiments adherent primary cultures (initially 30,000 cells seeded per 96 well plate or 100,000 cells per 24 well plate) were transfected with Lipofectamine 2000 reagent (Invitrogen), using 0.2 μg plasmid DNA and 0.5 μl Lipofectamin 2000 per 96 well plate. For colayer experiments bottom layer cells were initially seeded, either as 40,000 cells per 24 well plate (in vitro studies) or 400,000 cells per 6 well (for grafting experiments). On DIV4 sister cultures were detached and total cell number estimated. For nucleofection 2 million cells were resuspended in 100 μl nucleofection solution and transfected with 5 μg of plasmid DNA using the Amaxa basic nucleofector kit for primary neurons and the nucleofector program A-033 (12). Cells were either seeded on polyornithine/laminin-coated plates (standard protocol) or for colayer protocol on top of the bottom layer cultures in a certain ratio of 1:3, 1:6, or 1:8, respectively.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and further processed using standard protocols. The following primary antibodies were used: anti-FLAG-M2 (F1804, Sigma, 1:500), antiglial fibrillary acidic protein (GFAP) antibody (G3893, Sigma-Aldrich, 1:400), anti-nestin antibody (MAB-353, Chemicon, 1:600), anti-NeuN (MAB377, Millipore, 1:100), and anti-tyrosine hydroxylase (TH) antibody (MAB-5280, Millipore, 1:200 or AB152, Millipore, 1:1,000). The secondary anti-rabbit/mouse antibodies were conjugated with Alexa488, Alexa555 (A11034, A21424, A21429, Invitrogen, 1:500) or Cy3 (115-165-003, Jackson ImmunoResearch, 1:200). Nuclei were visualized by 4,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich, 1: 1,000) staining.
Cell Counting In Vitro
TH-ir cell number of standard cultures in 96-well plates were counted in pictures taken at 4x magnification from 6–10 randomly selected fields (each 2.4 mm2, corresponding to 13% of the total well surface) per independent experiments (n = 4–6). Green fluorescent pCAGGS-EGFP-transfected cells colocalizing with different cell markers (GFAP-ir, nestin-ir, NeuN-ir, or TH-ir) were counted at 20x magnification directly under an inverse Olympus fluorescent microscope (at least 100 EGFP+ cells per experiment, n = 3–4). TH-ir cell number of the densely colayer cultures (24-well plates, n = 4–6) were counted in two stripes (horizontally and vertically), corresponding to 4.5% of the total well surface, through the well at 20x magnification directly under an inverse Olympus fluorescent microscope. Total number of cells was determined in Neubauer counting chamber from detached sister cultures.
Transplantation of DA Cells
Eight weeks after 6-OHDA lesion, transplantation surgery was performed as described previously (44). Briefly, rats received four deposits (two medial and two lateral) of 1 μl cell suspension each (130,000 cells/μl in 0.05% DNAse/DMEM) into the right striatum at the coordinates (in millimetres with reference to bregma and dura, tooth bar 0): medial tract AP +1.0, LAT −2.3, VERT -5 and -4; lateral tract AP +1.0, LAT −3.3, VERT -5 and -4. Three sets of experiments were performed (details see below). The culture media used during the in vitro differentiation period are abbreviated (e.g., 1A, 1 day attachment medium; 3P, 3 days proliferation medium; 2D, 2 days differentiation medium).
In Vivo Experiment 1 (Different Cell Culture Protocols of EGFP Transfected Cells)
Eighteen lesioned rats were divided equally over three experimental groups. All animals received E12 ventral midbrain-derived cell suspensions, which had been cultured for 4 days (1A+3P). Then cells were detached and reseeded on empty plates (nontransfected control group), or detached and nucleofected with an EGFP expression plasmid and afterwards either seeded on coated empty plates (standard group) or on top of an adherent sister culture at a ratio of 1:8 (colayer group). After three additional days in culture (1A+2D) cells were used for transplantation (n = 6 animals per group). Two weeks later animals were sacrificed and brains were processed for immunohistochemistry (see below).
In Vivo Experiment 2 (Different Colayer Ratios of BDNF and EGFP Transfected Cells)
All experimental groups received cells cultured for 1A+3P, then were either nucleofected (BDNF and EGFP-transfected groups) or just detached (nontransfected group) and reseeded as 1:3 or 1:6 colayer for 2D in vitro. In the final analysis 38 lesioned and transplanted rats were included, which were allocated into five groups: nontransfected (colayer ratio 1:6, n = 9), EGFP-FLAG transfected (colayer ratio 1:6, n = 9 or 1:3, n = 7), and BDNF-FLAG transfected (colayer ratio 1:6, n = 7 or 1:3, n = 6). Rats were sacrificed two weeks after transplantation and brains were processed for immunohistochemistry (see below).
In Vivo Experiment 3 (Long-Term BDNF and EGFP Colayer Study)
In the final analysis 36 lesioned and transplanted rats were included, which were allocated into three groups: nontransfected (colayer ratio 1:3, n = 9), EGFP-FLAG transfected (colayer ratio 1:3, n = 14), and colayer BDNF-FLAG transfected (colayer ratio 1:3, n = 13). Cells were cultured for 1A+3P, then either nucleofected (BDNF and EGFP-transfected groups) or just detached (nontransfected group) and reseeded as 1:3 colayers for 2D in vitro. Drug-induced rotation behavior was assessed every third week. Animals were sacrificed 13 weeks after transplantation and brains were processed for immunohistochemistry (see below).
Histology and Immunohistochemistry
Two or 13 weeks after transplantation surgery, respectively, animals were deeply anesthetized with chloral hydrate and transcardially perfused with 125 ml 0.9% saline followed by 250 ml of 4% paraformaldehyde in PBS. Brains were postfixed overnight and cryoprotected by 30% sucrose treatment at 4°C. Coronal brain sections (between 3.2 and −7.8 mm according to bregma) were cut on a freezing microtome at 40 μm thickness in series of six. Cryoprotection of the sections was done at −80°C in antifreeze medium [30% glycerine (v/v), 30% ethylenglycol (v/v), 40% PBS]. Immunohistochemistry was performed on free-floating sections. Every third section was stained with anti-TH antibody (T1299, Sigma, 1:2500), visualized by avidine-biotincomplex (ABC) kit (Vector Labs) and 3′,3-diaminobenzidine (DAB) to allow stereological quantification of the surviving TH-ir cells. The remaining sections were processed for immunofluorescence using: anti-FLAG-M2 (F1804, Sigma, 1:500), anti-G-protein-gated inwardly rectifying K+ channel (Girk2; Kir3.2, APC-006, Alomone labs, 1:100), and anti-TH (AB152, Millipore, 1: 500 or T1299, Sigma, 1:500) antibodies. The secondary anti-rabbit/mouse antibodies were conjugated with Alexa488 (A11029, Invitrogen, 1:500), Alexa555 (A21429, Invitrogen, 1:750), and Cy2 (115-225-003, Jackson ImmunoResearch, 1:200).
Microscopic Analysis and Stereological Quantification
Confocal microscopy was performed on a Leica TCS SP2, equipped with Leica acquisition software, using oil immersion objective HCX PL APO BL (63x, numerical aperture 1.4) and HCX PL APO CS (40x, numerical aperture 1.25). DAB-stained TH-ir cells were quantified using the model-based (2D) cell counting method (4) using the optical CAST-grid system (Olympus) on an Olympus BX50 microscope equipped with a motorized stage. After the graft was outlined in every third section (120 μm apart) at 10x magnification, all TH-ir cells within the section profile were counted at 100x magnification (oil immersion, numerical aperture 1.25). The total number of TH-ir cells per graft was calculated using the formula of Abercrombie (1). The correction factor was calculated using cell diameter and tissue thickness measurements in the z-axis obtained with an electric microcator (ND 281; Heidenhain, Traunreut, Germany). The degree of morphological reinnervation was quantified by measuring the optical fiber density of TH-ir fiber outgrowth into the striatum using ImageJ software at four anterior–posterior sections in the central part of medial and lateral grafts separately. Distance of the TH-ir fiber outgrowth (TH-ir halo thickness) was measured perpendicular to the graft border at eight positions surrounding the graft, excluding fiber outgrowth in the region between medial and lateral graft when both halos were fused. For TH-ir fiber density measurements background value measured in the corpus callosum was subtracted from the striatal value. The results are given as mean in percentage of the contralateral intact side. All morphometric measurements were conducted in a blinded manner using coded sections. Images were assembled with Adobe Photoshop.
Statistical Analysis
Results are expressed as means ± SEM. Comparison between two groups were performed by Mann-Whitney U-test or if indicated separately by paired Student t-test. Two-way analysis of variance (ANOVA) followed by Bonferroni post hoc test was performed for rotation data, where type of transplant and time were two factors, using GraphPad Prism 4 software. Values below p < 0.05 were considered as statistically significant.
Results
Characterization of the pCAGGS Expression Plasmid
As expression levels of transfected genes depend strongly on the promoter and cell type used, we first compared the CMV promoter-based EGFP expression plasmid we used previously (12), with a CAG promoter-based expression plasmid pCAGGS-EGFP. Transfection of primary ventral mesencephalic cells with pCAGGS-EGFP resulted in higher numbers of EGFP+ cells, brighter fluorescent signal, and, importantly, long-lasting expression for at least 28 days in vitro, whereas CMV promoter-driven EGFP expression was barely detectable after 14 days (data not shown). Therefore, pCAGGS-derived plasmids were used for all subsequent experiments.
As dissociated cultures of the embryonic VM contain several cell types, we were interested if the CAG promoter was active in all three major cell types (progenitor cells, neurons, and astrocytes) and DA neurons (Fig. 1A–D). Two days after pCAGGS-EGFP transfection most of the green fluorescent cells (EGFP+) colocalized with nestin (59 ± 6%), a marker of progenitor cells, followed by 14 ± 3% NeuN-ir neurons and few GFAP-ir astrocytes (1 ± 1%) (n = 4). Six days after transfection the number of EGFP+/nestin-ir cells was reduced to 30 ± 5%, whereas the number of EGFP+/NeuN-ir (22 ± 2%) and EGFP+/GFAP-ir (15 ± 3%) was increased (n = 3). Differences between day 2 and day 6 values of each group did not reach statistical significance, although p-values for nestin and GFAP group, respectively, failed barely (p = 0.06, U-test). This shift in the proportion of immature progenitor cells (nestin-ir) towards mature neuronal (NeuN-ir) and glial (GFAP-ir) cell types reflects the ongoing differentiation process in these cultures and indicates that pCAGGS-EGFP-transfected cells are still able to differentiate (Fig. 1E). The number of EGFP+/TH-ir labeled cells was similar at both time points: 1.7 ± 0.7% and 1.5 ± 0.5%, respectively (Fig. 1E).
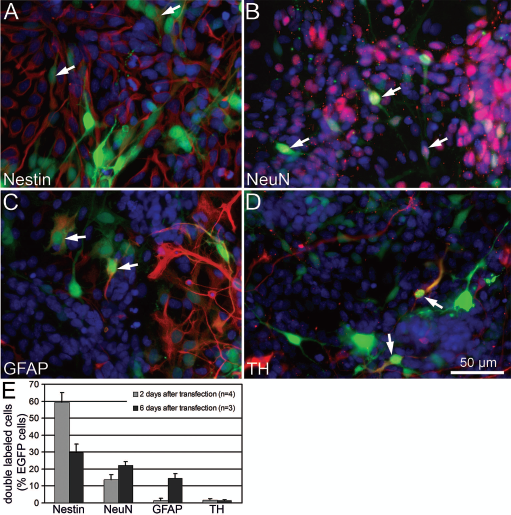
Characterization of the pCAGGS plasmid in primary ventral mesencephalon (VM) cell cultures. (A–D) CAG promoter-driven enhanced green fluorescent protein (EGFP) expression (green signal) was confirmed in four different cell types stained in red: (A) nestin-ir progenitor cells, (B) NeuN-ir neurons, (C) glial fibrillary acidic protein (GFAP)-ir astrocytes, and (D) tyrosine hydroxylase (TH)-ir neurons. Arrows point to EGFP-expressing cells colabeled with the respective cell marker. Cell nuclei were visualized by 4′,6-diamidino-2-phenylindole (DAPI) staining (blue signal). (E) The proportion of doubled labeled cells (cell marker-ir/EGFP+) among the total number of transfected green fluorescent cells (EGFP+) was quantified after pCAGGS-EGFP transfection of VM cell preparations cultured for 2 or 6 days in differentiation medium. The proportion of EGFP+/nestin-ir cells was decreased at day 6 after transfection compared to day 2, whereas the proportions of EGFP+/GFAP-ir cells and EGFP+/NeuN-ir cells were increasing.
Characterization of pCAGGS-BDNF-FLAG Expression Plasmid
BDNF was selected for biological validation of the pCAGGS expression plasmid, because of its known dopaminergic differentiation-enhancing effect (5,20,23, 29,31). The full-length rat BDNF coding sequence was inserted into pCAGGS and a 3xFLAG epitope tag (~3 kDa) was added at the C-terminus to allow sensitive and convenient detection of the BDNF-FLAG protein (Fig. 2A). EGFP-FLAG-transfected cells served as control (Fig. 2B). The BDNF protein is produced as 32-kDa precursor (proBDNF) and after secretion becomes proteolytically cleaved to yield the 13-kDa mature BDNF form (32,49), which correspond to 35 kDa proBDNF-FLAG and 16 kDa mature BDNF-FLAG with our 3xFLAG-tagged construct. An anti-BDNF antibody was used to compare the amount of BDNF-FLAG in the conditioned media (collected 2 days after transfection, 1:3 colayer culture) with a known standard of recombinant BDNF protein (Fig. 2C). Densitometry of the Western blots revealed an average of 16.5 ± 3.3 ng/ml (n = 4) mature BDNF-FLAG in the conditioned media. Incubation of primary ventral mesencephalic cells with conditioned media from pCAGGS-BDNF-FLAG-transfected cells for either 5 or 30 min resulted in phosphorylation of BDNF receptor TrkB, to a similar extent as if cells were incubated with recombinant mature BDNF (rBDNF, 20 ng/ml) (Fig. 2D). Thereby functionality of plasmid-based delivered BDNF-FLAG was confirmed. In contrast, conditioned media from pCAGGS-EGFP-FLAG-transfected cells did not induce TrkB phosphorylation (Fig. 2D). The effect of BDNF-FLAG overexpression on the differentiation process of DA neurons was studied on lipofectamine-transfected VM cell preparations (Fig. 2E). Transfection with pCAGGS-BDNF-FLAG resulted in 25 ± 8% increase of TH-ir neurons compared to pCAGGS-EGFP-FLAG-transfected cultures (p < 0.05, paired t-test, n = 6). pCAGGS-empty transfected cultures treated with 40 ng/ml rBDNF for 5–7 days serves as control, which resulted in 35 ± 8% increase in the number of TH-ir neurons (p < 0.005, paired t-test, n = 4) (Fig. 2E). No significant differences were observed between rBDNF treatment and BDNF-FLAG transfection or between EGFP-FLAG transfection and empty plasmid controls, respectively.

Overexpressed brain-derived neurotrophic factor (BDNF)-FLAG protein is biologically active and can be detected by immunocytochemistry or Western blot. (A, B) The C-terminal FLAG epitope allows detection of BDNF-FLAG (A) and EGFP-FLAG (B) by immunocytochemistry. (C) The conditioned media from pCAGGS-BDNF-FLAG-transfected (BM1–BM4) cells, but not from pCAGGS-EGFP-FLAG (EM1–EM4) transfections, contains the mature BDNF-FLAG (16 kDa) and BDNF-FLAG precursor (35 kDA). Two days after transfection (1:3 colayer, 24-well plate) 50 μl conditioned media from four independent experiments were analyzed by Western blot with anti-BDNF antibody; 2 ng recombinant BDNF (rB, 13 kDa) served as positive control. (D) Cell lysates (50 μg total protein) of nontransfected ventral mesencephalic cultures were analyzed by Western blot. The lower blot was probed with an anti-tyrosine kinase receptor B (TrkB) antibody, showing the 145-kDa full-length signal-transducing receptor TrkB and the 95-kDa C-terminal truncated TrkB receptor (without tyrosine kinase domain). The upper blot was probed with phospho-TrkB antibody (pTrkB, Tyr706/707). Only cells that were preincubated for 5 or 30 min with recombinant BDNF (rB, 20 ng/ml) or conditioned media from BDNF-FLAG-transfected cells (BM) show phosphorylation of full-length TrkB, whereas conditioned media from EGFP-FLAG-transfected cells (EM) did not induce this effect. (E) Transfection with pCAGGS-BDNF-FLAG or media supplementation with recombinant BDNF (40 ng/ml rBDNF) increases the number of TH-ir neurons by 25% or 35%, respectively, compared to pCAGGS-EGFP-FLAG transfections (*p < 0.05, **p < 0.005, paired t-test, n = 4–6). Primary VM preparations seeded in 96-well plates were transfected with lipofectamine on DIV4 (1A+3P), cultured in differentiation medium for 5–7 days, and stained with anti-TH antibody. TH-ir cell number after transfection with pCAGGS-empty was set to 100%.
The Colayer Method Maintains TH-ir Cell Number
In a previous study we have shown that nucleofection was the method of choice to transfect VM progenitor cells compared to lipofection or electroporation (12). However, we noticed that the number of TH-ir neurons was decreased in the nontransfected detached group and the EGFP-transfected group to 61% and 40%, respectively, compared to the nontransfected control (Fig. 3A). Possibly the nucleofection itself and/or the accompanied detachment and reseeding procedure may cause the loss of the vulnerable DA neurons. To circumvent this drawback we established the colayer method. Basically, freshly prepared VM cells are seeded into two separate wells. After identical culture conditions for 4 days, cells from the first well are detached and nucleofected as usual. Afterwards these cells, now referred to as top layer, were reseeded in certain ratios, such as 1:8, 1:6, or 1:3, on top of the unmodified sister culture in the second well (referred to as bottom layer). Quantification of TH-ir neuron number revealed that the colayer method assures similar numbers compared to nontransfected standard controls (Fig. 3A). In addition, TH-ir cells of the standard transfection displayed shorter outgrowths (Fig. 3B) compared to colayer cultures (Fig. 3C), due to the nondetached bottom layer TH-ir cells. As the standard nucleofected control (Fig. 3D) contained fewer cells in total (0.61 ± 0.09 million cells) compared to the colayer (2.24 ± 0.14 million cells) (Fig. 3E), quantification of TH-ir cell number (Fig. 3A) was corrected by the total number of DAPI-stained cells (see Materials and Methods). In both cases similar numbers of EGFP-transfected cells were seeded, either on coated empty plates (Fig. 3F) or as 1:3 ratio on top of the bottom layer cells (Fig. 3G). Interestingly, 2 days after transfection EGFP fluorescent cells displayed shorter neurites, when seeded on coated empty wells (Fig. 3H) compared to cells seeded on bottom layer cells (Fig. 3I), indicating a better differentiation under colayer conditions.

In vitro validation of the colayer setup. (A) Cell detachment and EGFP transfection reduces the number of TH-ir neurons to 40% (*p < 0.05), whereas EGFP transfection in 1:3 colayer setup maintains TH-ir number (*p < 0.05, n = 4). Number of TH-ir neurons was calculated in relation to the total cell number obtained from detached sister cultures counted in the Neubauer chamber. The TH-ir cell number of nontransfected cultures was set to 100%. (B–G) Four days after EGFP transfection cells reseeded on coated empty plates display fewer TH-ir cells with shorter outgrowths (B) compared to colayer cultures (C). This difference is mainly contributed by the bottom layer cells, which were never detached. Of note, the standard nucleofected control (D) contained fewer cells in total (blue DAPI staining) compared to the colayer (E), as identical numbers of EGFP-transfected cells (green signal) were seeded on both coated empty plates (F) and wells containing bottom layer cells, in a 1:3 ratio in the latter case (G). (H, I) EGFP-transfected cells seeded as colayer (I) display a more pronounced neuron-like outgrowth compared to standard cultures (H) 2 days after nucleofection. (J) Transfection with an BDNF-FLAG expression plasmid, which encodes for the secreted neurotrophic factors BDNF, in the 1:3 colayer setup increases the number of TH-ir neurons by 25%, compared to EGFP-FLAG transfection (*p < 0.05, paired t-test, n = 7). After transfection colayers were differentiated for 4–6 days and stained for TH. Scale bars: 200 μm (for B–G) and 100 μm (for H–I).
As the advantage of the high TH+ cell number is mainly contributed by the nontransfected/not detached bottom layer cells at the expense of a lower transfection rate (dilution of the transfected cells 1:3) we were interested if BDNF-FLAG transfection is still sufficient to influence DA neuron differentiation in this setup. Indeed, BDNF-FLAG-transfected 1:3 colayer cultures displayed an increased number of TH-ir neurons by 25 ± 7%, compared to EGFP-FLAG-transfected 1:3 colayers (p < 0.05, paired t-test, n = 7) or nontransfected 1:3 colayer controls, respectively (Fig. 3J).
First and Second Short-Term In Vivo Experiment
The influence of the in vitro pretreatment, such as detachment, nucleofection, or colayer, on the number of TH-ir neurons was analyzed 2 weeks after intrastriatal grafting into 6-OHDA-lesioned rat brains (n = 6 animals per group). The extensive TH-ir fiber network of the “healthy” contralateral striatum (Fig. 4A) is absent on the lesioned side containing the medial and lateral implants of embryonic DA neurons (Fig. 4B, C). At this stage the transplanted DA neurons have finished their first maturation phase (21) and extend few TH-ir axonal outgrowths into the host striatum (Fig. 4D, E). Morphometric analyzes revealed that animals receiving cells processed with the standard transfection protocol (all cells detached, nucleofected with pCAGGS-EGFP, and reseeded on new plates) displayed a significantly reduced TH-ir neuron number of 271 ± 62 compared to the 1,020 ± 173 TH-ir cells of the nontransfected control group (all cells detached and reseeded in a coated empty well, p < 0.005). In contrast the 1:8 colayer protocol resulted in higher numbers of TH-ir neurons (1,723 ± 199), which differed significantly compared to the control group (p < 0.05) and to the standard transfection group (p < 0.005) (Fig. 4F). Thus, similar to the in vitro data (Fig. 3A), detachment and more pronounced detachment plus nucleofection results also in vivo in a dramatic reduction of the number of transplanted TH-ir neurons to 59% or 16%, respectively (Fig. 4F). As the initial in vivo colayer ratio of 1:8 resulted in low numbers of transfected cells, we increased the proportion of nucleofected top layer cells to a ratio of 1:6 or 1:3, respectively. This time nontransfected 1:6 colayer served as control. Two weeks after intrastriatal transplantation no significant differences in the number of surviving TH-ir neurons were observed between BDNF-FLAG-transfected, EGFP-FLAG-transfected, and control groups, nor between 1:3 or 1:6 colayer ratios (Fig. 4G). Differences in the TH-ir cell number between the initial 1:8 colayer experiment (Fig. 4F) and the 1:3 and 1:6 colayer experiments (Fig. 4G) were not statistically significant.

In vivo validation of the colayer setup 2 weeks after transplantation. (A–C) The micrographs show coronal rat brain sections at the level of the caudate-putamen unit, which have been processed by TH immunohistochemistry with DAB visualization (brown staining). The nonlesioned striatum displays a dense network of TH-ir fibers (A), which is absent in the respective section of the6-hydroxydopamine (6-OHDA)-lesioned side (B). In the center of the lesioned striatum the medial and lateral graft are clearly distinguishable (B). The TH-ir cells and neurites are evenly distributed throughout the graft (C). (D, E) Confocal images of fluorescent-labeled TH-ir (red) and EGFP-FLAG-ir (green) cells and processes 2 weeks after transplantation, showing the border (dotted line) between graft (left) and host (right) tissue. The transfected EGFP-FLAG-ir cells and the TH-ir neurons are intermingled evenly throughout the graft at this stage (D). Few graft derived TH-ir neurons innervating the host striatum can be seen close to host–graft boundary (star in D marks the higher magnified region in (E). (F) The effect of the in vitro pretreatment on the number of intrastriatal transplanted TH-ir was quantified 2 weeks after surgery (n = 6 animals per group). Highest numbers of grafted TH-ir neurons were found in the EGFP-transfected 1:8 colayer group, whereas this number was reduced to 16% in the EGFP-transfected standard group (**p < 0.005). The control group, which received cells that were detached but not transfected, contained an intermediate amount of TH-ir cells (59% compared to the colayer group, *p < 0.05), indicating that the detachment and reseeding procedure, which is also part of nucleofection protocol, accounts partly for the decreased number of TH-ir neurons. (G) Varying the ratio of EGFP-transfected top layer cells to nontransfected bottom layer cells, 1:3 or 1:6, respectively, had no effect on the total number of TH-ir neurons 2 weeks after grafting and was similar to nontransfected 1:6 colayer controls. In contrast to the expectations, neither 1:6 nor 1:3 BDNF-transfected colayers displayed an increased number of TH-ir neurons in vivo (no significant differences between groups, n = 6–9 animals per group; see Materials and Methods for details).
Third In Vivo Experiment (13 Weeks Long Term)
As time point-dependent effects of rBDNF applied on DA grafts by minipumps into the striatum close to the transplantation site have been described (50), we investigated BDNF-FLAG-transfected colayer grafts over a 12-week period in vivo by monitoring the drug-induced rotation behavior every third week. Generally, functional integration of DA grafts into the host basal ganglia circuitry is characteristic for the second phase of the implant integration process (21). Amphetamine-induced ipsilateral rotation behavior decreased in all three colayer groups (nontransfected, EGFP transfected, and BDNF transfected) similarly, from initial pregrafting values of 13.0, 14.8, and 13.2 rounds per minutes, respectively, to 3.1, 3.6, and 1.5 rounds per minutes 12 weeks after transplantation, reaching statistical significance already 3 weeks after grafting in each group (Fig. 5). However, two-way ANOVA analysis of amphetamine-induced rotation data revealed that the type of transplant did not affect the results (p > 0.05). Apomorphine-induced contralateral rotation behavior decreased from initial pregrafting values of 12.5 rounds per minute to 8.6 (nontransfected), 6.7 (EGFP transfected), and 7.7 (EGFP transfected) rounds per minute (Fig. 5B). Although, significant improvement of the rotation behavior of the nontransfected group was not observed until the ninth week after transplantation, overall comparison of the apomorphine-induced rotation data using two-way ANOVA analysis did not reveal an effect of the type of transplant (p > 0.05). In summary, transplantation of VM cells, which were previously in vitro cultured as 1: 3 colayer, improved drug-induced rotation behavior independent of the type of transplant. Neither did the transfection procedure using EGFP control plasmid impair rotational behavior compared to nontransfected colayer cells, nor did transfection with BDNF plasmid improve rotational behavior.

Rotational behavior and histology 13 weeks after transplantation. After administration of either amphetamine (A) or apomorphine (B) rotational behavior was monitored pregrafting (pg) and every third week (w) after grafting (3w, 6w, 9w, 12w) for three experimental groups, which received either native (black bars, n = 9), EGFP-transfected (gray bars, n = 14), or BDNF-transfected (white bars, n = 13) 1:3 colayer transplants. (A) Amphetamine-induced ipsilateral rotation declined in all three groups similarly, reaching statistical significance already 3 weeks after grafting. (B) Apomorphine-induced rotational behavior reached statistical significance 9 weeks after transplantation in the native transplant group, whereas in the EGFP- or BDNF-transfected group, respectively, significance level was reached already 3 weeks after grafting (two-way ANOVA followed by Bonferroni post hoc test, ns = not significant, **p < 0.01, #p < 0.001, compared to the pregrafting scores of the same experimental group) (C, D) Thirteen weeks after transplantation the striatal reinnervation extended much further from the graft–host border and the graft volume increased, compared to the situation after 2 weeks (compare Fig. 4). (E) At higher magnification a dense network of TH-ir fibers (red) innervating the host striatum are visible. (F, G) In 13-week-old grafts the majority of the TH-ir neurons (red) reside primarily at the periphery of the graft (F) and extend a dense network of TH-ir fibers into the surrounding host striatum (G, dotted line marks the graft–host border, star marks higher magnified region in E). In contrast the EGFP-FLAG-ir (green) cells and fibers remained in the center of the graft (F), similar to that observed after 2 weeks, and rarely extended processes into the host tissue. (H) Double labeling for TH-ir (green) and G-protein-gated inwardly rectifying K+ channel (Girk2-ir; red) (yellow in the merged image, arrowheads) revealed the presence of typical A9 (substantia nigra pars compacta) derived embryonic midbrain dopamine (DA) neurons in the grafts close to the host border.
Thirteen weeks after grafting the total number of TH-ir cells in the transplants were quantified on DAB-stained sections (Fig. 5C, D). However, no differences between nontransfected, EGFP-FLAG, and BDNF-FLAG groups were identified in the following parameters (Table 1): TH-ir neuron number, graft volume, TH-ir neuron density, TH-ir fiber density, and extend of the TH-ir fiber outgrowth (thickness of the TH-ir halo around the graft). The prominent TH-ir halo at this time point reflects more intense reinnervation of the host striatum (Fig. 5C) compared to the 2-week groups (Fig. 4B). This difference can be clearly seen in confocal images at high-power magnification (compare Figs. 4E and 5E). Additionally, 13 weeks after transplantation DA neurons are preferentially located close to the graft–host boundary (Fig. 5F), in contrast to an even distribution throughout the graft seen 2 weeks after transplantation (Fig. 4C, D). Such redistribution was not seen for EGFP-FLAG-transfected neurons, which remained uniformly scattered throughout the transplants at both time points (Figs. 4D, 5F). EGFP-FLAG-positive neurites were rarely detected in the surrounding host striatum; instead the EGFP-FLAG-ir fiber networks remained within the transplant (Fig. 5F, G). Due to the dissection procedure of the embryonic midbrain two subtypes of DA neurons, which localized either in the substantia nigra pars compacta (A9 region) or ventral tegmental area (A10 region), are normally present in the transplants. Notably, the A9 subpopulation has critical importance to improve motor performance, as recently demonstrated by grafting of VM preparations of Pitx3-deficient mice, which failed to develop A9 neurons, compared to wild-type grafts containing both A9 and A10 DA neurons (18). Therefore, we verified proper differentiation of the grafted cells into A9 subtype of DA neurons by Girk2-ir/TH-ir double staining (Fig. 5H). Preliminary quantification of the 13-week transplants revealed that the majority (70–80%) of DA neurons were double labeled for Girk2-ir/TH-ir, which is in good agreement with previously reported data (18).
Morphometric Quantification of the Intrastriatal Grafts After 13 Weeks

Thirteen weeks after transplantation graft morphology was quantified in several parameters after sections were stained with 3,3′-diaminobenzidine (DAB) for tyrosine hydroxylase immunoreactivity (TH-ir). No significant differences between the three 1:3 colayer groups [nontransfected, enhanced green fluorescent protein (EGFP)-FLAG transfected, and brain-derived growth factor (BDNF)-FLAG transfected] were observed.
Discussion
The lost DA input in the PD striatum can be partially replaced by intrastriatal transplantation of DA neurons, which results in behavioral improvements. However, the success of the restorative approach is limited by the low survival rate of the transplanted DA neurons (11). Among other attempts, cotransplantation of DA neurons and neurotrophic factor-producing cells (e.g., bFGF-2-secreting fibroblasts or Schwann cells) has been successfully applied to improve the transplantation outcome (42,45). The aim of the present study was to establish a protocol that enables efficient nonviral genetic modification of the transplanted cells, without decreasing the numbers of DA neurons in the graft. This can be achieved with the colayer method, where nucleofected cells are reseeded on top of nondetached sister cultures. Although the colayer method results in a 1:3 dilution of transfected cells, this limitation might be preferable over a strong reduction of DA neuron number to 40% in vitro or to 16% in vivo, when all cells underwent the nucleofection procedure. Another modification from our previous nucleofection procedure (12) was the use of a CAG promoter-containing expression plasmid (instead of CMV promoter), which ensured a strong and long-lasting gene expression for up to 13 weeks after transplantation in vivo. Similar to our data from transiently transfected rat NPCs, a prolonged EGFP expression for up to 10 weeks has been reported in stably transfected human neural progenitor cells containing a CAG promoter construct, whereas CMV promoter-based EGFP expression lasted only 6 weeks (13). Furthermore, as the CAG promoter was active in nestin-ir progenitor cells and neurons derived from embryonic VM cell preparations, this promoter is suitable to guide differentiation of the transfected cells towards DA neurons by overexpression of adequate transcription factors, such as the nuclear receptor Nurr1 (personal observation).
In contrast to transcription factors, secreted neurotrophic factors are excellent candidates for the colayer method, as it is not necessary to transfect the DA neurons directly; instead the extracellularly released factors can reach neighboring nontransfected DA cells indirectly. Exogenous delivered BDNF in particular is known to increase the number of DA neurons in vitro (23,29) and was therefore selected for this initial colayer study. However, BDNF-transfected colayers produced ambivalent results during the in vitro and in vivo validation of the colayer method. In vitro BDNF-FLAG-transfected colayers contained 25% more DA neurons compared to nontransfected controls. Despite these promising in vitro results, no differences were observed after in vivo application of BDNF-FLAG-transfected colayer grafts when compared to the EGFP-FLAG-transfected or nontransfected control groups. Taking into account that the majority (80–99%) of the transplanted DA neurons do not survive the transplantation procedure (11), our results indicate that BDNF was not sufficient and/or adequate to enhance the survival of the transplanted DA neurons. In agreement to that, it has been reported previously that daily intrastriatal injections of mature BDNF protein next to grafted DA neurons did not increase DA neuron survival (40). Moreover, it has been shown that the effect of BDNF depends on the time course of BDNF delivery. Yurek et al. reported that intrastriatal infusions of mature BDNF protein showed optimal effectiveness, if animals received BDNF in the third and fourth week after transplantation of DA neurons rather than in the first and second week (50). Therefore, it is possible that the continuous release of BDNF by the transfected colayer cells was not optimal to improve TH-ir fiber outgrowth. Although BDNF promotes differentiation of DA neurons in vitro and protects these cells against 6-OHDA and MPTP toxicity in vivo (3,23,29,46), up to now the effects on grafted DA neuron survival after in vivo administration, including the present study, are not promising (22,40,50–52).
In contrast to treatments with recombinant mature BDNF protein, the situation of full-length BDNF expression constructs, like the BDNF-FLAG we used, might be further complicated by the fact that in addition to the mature BDNF isoform the proBDNF precursor is also expressed, which is known to induce apoptosis mediated by p75 neurotrophin receptor (p75NTR) (28,43). As both TrkB and p75NTR receptors have been identified on DA neurons (8,36), mature BDNF and proBDNF might influence these cells directly. Furthermore, intrastrital grafting of a hippocampal neuronal stem cell line HiB5, which had been engineered and selected for stable BDNF expression (both isoforms), provoked a marked atrophy of the striatal formation 3 months after transplantation (39). In another cell line, C17.2, derived from the cerebellum, stable expression of BDNF prevented even neuronal differentiation of these cells (39). Therefore, adjustable expression systems such as tetracycline-inducible promoters are required to separate early graft survival-promoting effects from potentially later occurring deleterious effects.
In conclusion, the colayer method provides an efficient way for nonviral transfection without reducing the DA neuron number in the graft. Although BDNF was insufficient to enhance the survival of the transplanted DA neurons, the colayer method itself, with its possibility to apply differentiation and/or survival-promoting factors directly within the grafts, will be helpful to discover such factors or factor combinations in the future.
Footnotes
Acknowledgments
This work was supported by Georg-Christoph-Lichtenberg scholarships (I.K.) provided by the Ministry of Science and Culture of Lower Saxony and by EU Marie Curie host fellowships (A.N.) for early stage researchers training - MEST-CT-2005-021 014. We thank Dr. Hitoshi Niwa and Dr. James R. Whiteford for providing the pCAGGS-empty and pCAGGS-EGFP plasmids. We are grateful to Prof. Guido Nikkhah for introduction to stereological quantification and helpful discussions, as well as to Prof. Thomas Brinker for providing the CASTgrid microscope system. We thank Prof. Peter Claus for acquiring the confocal images. The excellent technical assistance of Silke Fischer, Natascha Heidrich, and Kerstin Kuhlemann is gratefully acknowledged. The authors declare no conflict of interest.