
Abstract
The differentiation of dopamine-producing neurons from chromaffin progenitors might represent a new valuable source for replacement therapies in Parkinson's disease. However, characterization of their differentiation potential is an important prerequisite for efficient engraftment. Based on our previous studies on isolation and characterization of chromaffin progenitors from adult adrenals, this study investigates their potential to produce dopaminergic neurons and means to enhance their dopaminergic differentiation. Chromaffin progenitors grown in sphere culture showed an increased expression of nestin and Mash1, indicating an increase of the progenitor subset. Proneurogenic culture conditions induced the differentiation into neurons positive for neural markers β-III-tubulin, MAP2, and TH accompanied by a decrease of Mash1 and nestin. Furthermore, Notch2 expression decreased concomitantly with a downregulation of downstream effectors Hes1 and Hes5 responsible for self-renewal and proliferation maintenance of progenitor cells. Chromaffin progenitor-derived neurons secreted dopamine upon stimulation by potassium. Strikingly, treatment of differentiating cells with retinoic and ascorbic acid resulted in a twofold increase of dopamine secretion while norepinephrine and epinephrine were decreased. Initiation of dopamine synthesis and neural maturation is controlled by Pitx3 and Nurr1. Both Pitx3 and Nurr1 were identified in differentiating chromaffin progenitors. Along with the gained dopaminergic function, electrophysiology revealed features of mature neurons, such as sodium channels and the capability to fire multiple action potentials. In summary, this study elucidates the capacity of chromaffin progenitor cells to generate functional dopaminergic neurons, indicating their potential use in cell replacement therapies.
Keywords
Introduction
Chromaffin cells in the adrenal medulla originate from the neural crest and, together with the sympathetic neurons of the dorsal ganglia and the intermediate small intensely fluorescent cells, constitute the sympathoadrenal (SA) lineage of neural crest derivates (32). During early embryogenesis, chromaffin cells and sympathetic neurons acquire their specific characteristics along their migratory route (38). Development from multipotent neural crest cells is regulated by distinct networks of transcription factors that specify sympathetic and adrenal chromaffin cell differentiation (50). These two SA cell lineages have in common catecholamine synthesis, including the enzymes tyrosine hydroxylase (TH) and dopamine-β-hydroxylase (DBH), as well as catecholamine storage and uptake.
In contrast to mature sympathetic neurons, chromaffin cells contain large dense-cored vesicles, express the epinephrine-synthesizing enzyme phenylethanolamine N-methyltransferase (PNMT), and lack neurofilament and neuritic processes in vivo (23,32). Furthermore, unlike sympathetic neurons, cells from adrenal medulla are able to renew throughout life (68,70).
The relatively high turnover of chromaffin cells suggests the existence of SA progenitor cells within the adrenal medulla. Recently, we established from bovine adrenal medulla the in vitro enrichment of chromaffin progenitor cells in spheres subjected to selective proneural culture conditions; these cells have the capability to self-renew and form clonal secondary spheres (10). In analogy to neurospheres, these floating clusters were termed chromospheres (10). Cellular subsets in these spheroid clusters express increased levels of neural progenitor markers such as nestin, Mash1, vimentin, Musashi1, and nerve growth factor receptor as well as markers of neural crest progenitor cells such as Sox1 and Sox10 (sex-determining region Y box 1 and 10) when compared to chromaffin cells. On the other hand, characteristics of differentiated chromaffin cells, such as the expression of PNMT, were reduced (10,16). The observed expression changes suggest the pronounced selection for SA progenitor's subset in vitro. These cells were capable to differentiate into neurons that express TH, the key enzyme in the synthesis of catecholamines. The capacity to differentiate into neurons suggests that isolated chromaffin progenitors may have therapeutic potential in the treatment of neurodegenerative diseases including the replacement of lost dopaminergic neurons in Parkinson's disease (15).
The close relation of chromaffin cells to catecholaminergic neurons has early on initiated trials to use these cells in the treatment of neurodegenerative diseases, and a substantial number of Parkinson patients had received autologous adrenal transplants from 1988 to 2001 with an encouraging improvement of clinical symptoms (12). A serious limitation in the application of adult adrenal medulla, however, is the postmitotic nature of most cells. Furthermore, it has to be considered that, in the adult adrenal, the proportion of dopaminergic cells is low (1% of the cells) while the vast majority of cells produces epinephrine. Therefore, in the present study, we aimed to establish protocols for the efficient and enhanced differentiation as well as enrichment of dopaminergic neurons from chromaffin progenitor cells.
Recent reports indicate that retinoic acid (RA) is a strong inducer of progenitor/stem cell differentiation (2), specifically of neural differentiation from human embryonic stem cells, hindbrain, and spinal cord progenitor cells (6,19,45,67). Furthermore, different lines of evidence suggest that RA is a potent inducer of dopaminergic differentiation of embryonic stem cells (19,60). In particular, RA was considered to have a main role in the induction of dopaminergic differentiation of adult neural progenitor/stem cells during regeneration (42,45). RA molecule plays an important role in the control of neural growth and patterning (42). Recent in vitro data suggest that ascorbic acid (AA) may increase the number of dopaminergic neurons by stimulating differentiation of neural precursors (4,61,74). Therefore, we tested the potential effects of RA and AA treatment in enhancing the capacity of multipotent SA progenitors to generate functional dopaminergic neurons.
Mastering the isolation of chromaffin progenitor cells and controlling their differentiation will open new avenues for regenerative therapies (14,22,63). Currently, cell-based therapies of neurodegenerative diseases are not practical due to a shortage of organ donors, lack of tissue homogeneity, and expandable cells besides disappointing survival rates of grafted cells (21). This study, together with our previous report, contributes to elucidating the capacity of chromaffin progenitors to derive functional neurons and provides a new insight into tools that may improve differentiation of these cells into specific neural lineages.
Materials and Methods
Isolation of Chromaffin Cells and Culture of Chromospheres
Bovine adrenal glands were obtained from freshly slaughtered 1- to 3-year-old cattles. Prior to cell isolation, adrenals were shortly put into 70% ethanol, and connective tissue was removed. To remove the remaining blood, the intact adrenals were flushed several times with PBS through the central vein. Primary chromaffin cells were isolated as previously described by collagenase and DNase digestion (25,62) (0.3% type II collagenase and 30 units/ml DNase I, both from Sigma-Aldrich, St. Louis, MO, USA). Cells from the adrenal medulla were separated by mechanical dissociation, sieved through 100-μm cell strainers (Becton Dickinson, Heidelberg, Germany), and washed with PBS. Primary chromaffin cells were then cultured in Dulbecco's modified Eagle's medium (DMEM)/F12 (Gibco, Invitrogen, Germany) supplemented with 10% steroid-free fetal bovine serum (FBS, Charcoal–Dextran treated, Hyclone, Logan, UT, USA) and 1% penicillin–streptomycin solution (Gibco) at 37°C in a humidified atmosphere (95% air, 5% CO2) overnight. Finally, chromaffin cells were separated from nonchromaffin cells by differential plating, based on the fact that nonchromaffin cells adhere faster than chromaffin cells. The cell suspension was transferred to a new culture flask once for 2 h and additional two times for 1 h. In our hands, the purity of the isolated medullary chromaffin cells is 97.5% (10,25). The isolated cells were seeded in neurobasal medium (Gibco) containing 2% B27-supplement (Gibco), 1% penicillin–streptomycin, 2 mM l-glutamine (PAA Laboratories, Cölbe, Germany), 20 ng/ml epidermal growth factor (EGF), 20 ng/ml bovine fibroblast growth factor (bFGF), and 10 ng/ml leukemia inhibitory factor (all Sigma-Aldrich) in ultra low attachment culture flasks (Sarstedt, Nümbrecht, Germany) where cells formed nonadherent spheroid clusters—chromospheres. Fresh medium was added every second day, and chromospheres were passaged weekly.
Limiting Dilution Analysis
For limiting dilution analysis, chromospheres from 1-, 2-, and 4-week-old cell cultures were treated with AccuMax (PAA Laboratories) to obtain single cell suspensions; cells were counted following staining with trypan blue (Sigma-Aldrich). Graded cell numbers were seeded into 96-well plates (1,000–8 cells/well) by plating recurring 1/2 dilutions. The presence of single cells per well was confirmed by microscopy. Seven days after seeding, wells were supplemented with fresh medium and growth factors. Sphere-positive wells were counted 14 days postseeding.
Estimation of Proliferation Capacity
Proliferation capacity was estimated using CellTrace™ CFSE Cell Proliferation Kit (Molecular Probes™, Invitrogen, Germany). Chromospheres were dissociated with Accumax (PAA Laboratories), and single cells were incubated with 10-μM carboxyfluorescein diacetate succinimidyl ester (CFDA-SE) in fresh media for 10 min at 37°C. Colorless CFDA-SE passively diffuses into cells where cleavage of its acetate groups by intracellular esterases converts the molecule to the fluorescent carboxyfluorescein succinimidyl ester (CFSE). The succinimidyl ester group covalently binds intracellular amines forming fluorescent conjugates. Cells were pelleted (5 min at 1,000 rpm) and resuspended in fresh media to release unconjugated CFSE reagent for 10 min at 37°C and additionally centrifuged. Proliferation rate was assessed by decreasing intensity of fluorescence at days 0, 3, 7, 12, 15, 17, and 21. Cells were analyzed on a FACS Calibur flow cytometer (BD Biosciences, USA) using a 488-nm argon ion laser and FL1 530/30-nm band pass filter for CFSE detection. Acquisition was performed by using CellQuest software (BD Biosciences).
Differentiation of Chromaffin Cells
For differentiation of chromosphere cells into neurons, 7 × 104 chromosphere single cells were plated onto 13-mm glass cover slips coated with poly-l-lysine (4 μg/ ml) and laminin (5 μg/ml) (PLL/L, Sigma-Aldrich). Cells were expanded for 2 days in neurobasal culture medium. Then differentiation of chromaffin progenitor cells was induced by a 3-day cultivation in differentiation medium I (DMEM/F12 supplemented with 1% N2 supplement (PAA Laboratories), 5 mM HEPES (PAA Laboratories), 1% penicillin–streptomycin solution, 2 mM l-glutamine, 7.5% sodium bicarbonate (PAA Laboratories), 5 μg/ml heparin (Sigma-Aldrich), 20 ng/ml EGF, 5 μM RA (Sigma-Aldrich), and 10 μM AA (Sigma-Aldrich). Cells were allowed to differentiate for another 3 days in differentiation medium II (DMEM/F12 supplemented with 2% B27 supplement, 5 mM HEPES, 1% penicillin–streptomycin, 2 mM l-glutamine, 7.5% sodium bicarbonate, 5 μg/ml heparin, 5 μM RA, and 10 μM AA). Growth factor was withdrawn in the second differentiation step.
Immunocytochemistry
Cells were immunostained with specific antibodies for the identification of dopaminergic neurons. The cells were fixed on slides with 4% paraformaldehyde in PBS for 10 min at 4°C, rinsed twice with cold PBS, and rendered permeable with 0.3% Triton X-100 in PBS [containing 5% goat serum and 1% bovine serum albumin (BSA)] for 1 h. Afterwards, cells were incubated with primary antibodies overnight at 4°C. Neurons were identified with rabbit polyclonal antibodies against β-III-tubulin (1:4,000, Convance, USA) or mouse monoclonal antibodies against TH (1:500, Chemicon) or microtubule-associated protein 2 (MAP2) (1:500, Chemicon). The cells were washed three times with PBS and incubated with the appropriate secondary antibody at room temperature for 2 h [goat anti-mouse IgG conjugated with cyanine 3 (Cy3), goat anti-rabbit conjugated with Cy3, goat anti-mouse conjugated with Cy2, Jackson Immuno Research]. Nonspecific background signal was evaluated by staining with secondary antibodies only. Cell nuclei were counterstained with DAPI (1:10,000 in PBS; 4′-6-diamidino-2-phenylindole, Sigma-Aldrich) for 5 s. After washing in PBS, slides were mounted in Aqua Poly Mount (Polysciences Europe, Eppelheim, Germany) and examined by fluorescence microscopy (Axioplan, Carl Zeiss, Jena, Germany). Percentages of differentiated neurons were calculated by counting total cell numbers based on DAPI staining and cell numbers of marker-positive cells. Analyses were performed using Axiovision software (Carl Zeiss).
Evaluation of Gene Expression Levels
Potential differences in gene expression between undifferentiated and differentiated cells were determined by reverse transcription-polymerase chain reaction (RT-PCR). Total RNA was isolated using RNeasy Plus Mini kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's instructions. One microgram of purified RNA was used for reverse transcription with the reverse transcription buffer, RNase inhibitor, oligo(dT)15 primer, dNTP mix, and M-MLV reverse transcriptase at 42°C for 60 min according to the manufacturer's specifications (Promega, Madison, WI, USA). PCR reactions were carried out with initial denaturation at 94°C for 5 min, annealing for 15 s, and elongation at 72°C for 15 s to 1 min (according to the expected product sizes), followed by final extension at 72°C for 1 min for qualitative analysis. Amplified PCR products were separated on 1% agarose gels, visualized with ethidium bromide, and documented (Gene Genius documentation system, Gene snap, Syngene, Cambridge, UK). Primer sequences and respective annealing temperatures are listed in Table 1.
Sequences and Annealing Temperatures of PCR Primers
Pax6, paired box protein 6; Hes1, hairy enhancer of split 5; MAP2, microtubule-associated protein 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; TH, tyrosine hydroxylase; Sox1, sex-determining region Y box 1; PNMT, phenylethanolamine N-methyltransferase; Pitx3, paired-like homeodomain 3; Nurr1, nuclear receptor-related protein 1; GTPCH, guanosine triphosphate cyclohydrolase.
Quantitative RT-PCR Analysis of Progenitor and Neural Markers
Standard curve for real-time PCR quantification was generated by amplification of DNA fragments for Mash1, nestin, paired-like homeodomain 3 (Pitx3), nuclear receptor-related protein 1 (Nurr1), guanosine triphosphate cyclohydrolase (GTPCH), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Table 1). PCR products were cloned into plasmid vectors (pCRIITOPO) and transformed into competent E. coli using the TOPO TA cloning kit (Invitrogen). Plasmids were purified using a Maxi Plasmid kit (Qiagen). Concentrations of plasmids were measured by spectroscopy, and a linear regression standard curve was created by serial dilutions of plasmids. PCR reaction contained 2 μl of cDNA sample, 5 μl of SYBR Green PCR master mix (Qiagen), 5 pmol of sense primer, and 5 pmol of antisense primer and proceeded on a Light Cycler 1.5 (Roche, Basel, Switzerland). Expression levels of Mash1, nestin, Pitx3, and Nurr1 gene markers were calculated by relative quantification to the expression of the reference gene GAPDH.
Determination of Catecholamine Release
Catecholamine release from differentiated bovine chromosphere cells was measured by liquid chromatography with colorimetric detection as previously described (17). Cultures incubated at 37°C with 400 μl/well extracellular solution (in mM: 124 NaCl, 3.25 KCl, 2 MgCl2, 2 CaCl2, 11 d-glucose, 10 HEPES, 1 AA, pH 7.4) were used as control. Release of catecholamines was stimulated by incubating cells with 56 mM KCl in extracellular solution (in mM: 71.25 NaCl, 56 KCl, 2 MgCl2, 2 CaCl2, 11 d-glucose, 10 HEPES, 1 AA, pH 7.4) (66). Upon 30-min stimulation, incubation media from three wells were collected, pooled, and stabilized by addition of 0.2 M acetic acid and stored at −80°C. Catecholamine release from cells incubated with RA/AA was compared to values obtained from control cells differentiated by standard procedure without addition of RA/AA. Catecholamine measurements were normalized to 10,000 cells.
Electrophysiological Recordings
Untreated and RA/AA-treated differentiated cells were investigated 8 days after initiating differentiation using standard whole-cell patch-clamp techniques at room temperature. Six hours prior to experiments, cells were incubated with 5% FBS to provide better sealing.
Extracellular solution contained (in mM) 142 NaCl, 8.1 KCl, 1 CaCl2, 6 MgCl2, 10 HEPES, 10 d-glucose, pH 7.4, and pipette solution contained (in mM) 153 KCl, 1 MgCl2, 5 EGTA, 10 HEPES, pH 7.3. Using these solutions, filamented borosilicate pipettes had resistances of 4–6 MΩ. Seal resistances in the whole-cell mode were between 0.1 and 1 GΩ. Resting membrane potentials (RMP) were determined immediately after gaining whole-cell access. Recordings were made in the whole-cell voltage-clamp or current-clamp mode, and data were recorded using an Axopatch 200B amplifier and Iso2 data acquisition software (Axon Instruments, Union City, CA, USA) essentially as described previously (28,65). Action potentials were elicited by applying increasing depolarizing current pulses (5 pA current steps). Analyzed cells were chosen based on morphological analogy to neurons.
Statistical Analysis
Quantitative data are expressed as mean ± standard deviation, and statistical significance was determined through one-way analysis of variance (ANOVA) with the post hoc Bonferroni's multiple comparison test or two-tailed Student's t test where appropriate. A value of p < 0.05 was considered significant in all tests.
Results
Chromosphere Initiation Capacity and In Vitro Differentiation of Chromosphere Cells
Similar to neurospheres, freshly isolated chromaffin progenitor cells form floating spherical clusters when cultured under low attachment conditions. These spherical clusters are heterogeneous structures containing different cell types including progenitor cells. Heterogeneity of spherical clusters undergoes changes during 21 days of culturing in proneural stem cell conditions, favoring growth of progenitor cells. This is reflected by a several folds of significant increase in the expression of the progenitor marker gene Mash1 (p < 0.05) (Fig. 1) indicating an increase of the progenitor subset within the spheres.

Mash1 progenitor marker gene expression. Gene expression was analyzed by quantitative real-time PCR in chromospheres up to 21 days of culturing. Data from three independent isolations were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. Displayed are mean values of fold change in Mash1 expression ± standard deviation (n = 3) (∗p < 0.05).
Self-renewal in vitro is defined as the capacity of cells from primary spheres to form secondary spheres (39). Our recent data provide evidence that chromosphere cells have the capability to self-renew and form clonal secondary spheres (10). To quantify chromosphere initiation capacity, graded numbers of enzymatically singularized cells from primary chromosphere were seeded into 96-well plates at 1, 2, and 4 weeks of culture. Frequencies of wells containing secondary spheres were recorded after 14 days. As shown in Figure 2A, the frequency of initiated secondary chromospheres was maintained within 2 weeks of culture. Chromosphere proliferation capacity was maintained for at least 4 weeks (ANOVA test, p > 0.05 with post hoc the Bonferroni's multiple comparison test, p > 0.05; 1, 2, and 4 weeks groups were compared) (Fig. 2A). In parallel, the reduction of proliferation rate was detected from days 15 to 21 of culturing using the CFSE assay (Fig. 2B). The same tendency was observed in all three chromosphere cultures analyzed. Therefore, 1-week-old cells, in pronounced proliferative phase, were used in further experiments.

Chromosphere initiating frequencies and proliferation analysis in chromosphere progenitor cultures. (A) Chromosphere initiating frequencies analysis after 1, 2, and 4 weeks culturing (analysis of variance, ANOVA, p > 0.05, n = 3). Single chromosphere cells (1,000–8 cells) were seeded, and sphere-positive wells were quantified. A representative analysis is shown. Insert shows chromospheres cultured for 2 weeks (original magnifications: 100x). (B) Proliferation assay after 0, 3, 7, 12, 15, 17, and 21 days (n = 3).
Analysis of Progenitor and Neural Marker Gene Expression
Expression of 14 progenitor and neural gene markers was analyzed by RT-PCR in 1-week-old chromosphere and in chromosphere cells after induction of neural differentiation. Progenitor markers, such as Sox1, Sox10, and Mash1, were downregulated during differentiation (Fig. 3A, left). Downregulation of Notch was accompanied by a decrease of downstream effectors basic helix-loop-helix (bHLH) gene repressors hairy enhancer of split 1 (Hes1) and Hes5 (Fig. 3A, left), key factors of maintenance of an undifferentiated state. Quantitative real-time PCR revealed significant downregulation of Mash1 upon neural differentiation (p < 0.05) (Fig. 3B). In contrast, the immature neural marker β-III-tubulin, the mature neural marker MAP2, and paired box gene 6 (Pax6) were upregulated (Fig. 3A, right). Expression of dopamine synthesizing enzyme TH was increased in differentiated cells versus chromosphere cells, while PNMT expression was reduced in selective differentiation conditions.
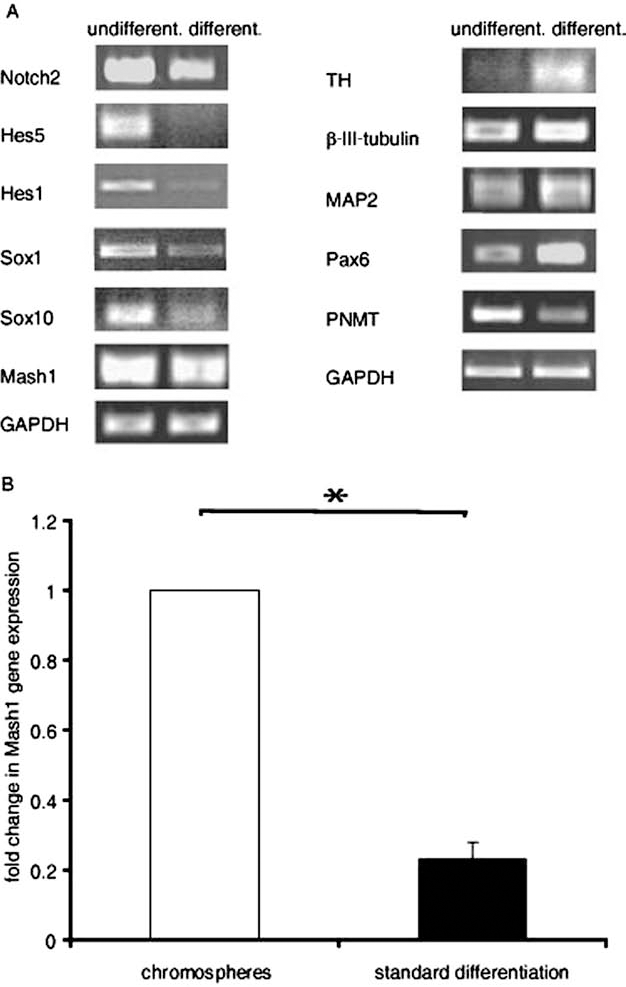
Gene expression analysis of undifferentiated and differentiated chromaffin progenitor cells. (A) mRNA expression of progenitor markers (left) and neural markers (right) of undifferentiated and differentiated chromosphere cells. GAPDH served as loading control. A representative analysis is shown. (B) Quantitative RT-PCR mRNA expression analysis of Mash1 neural progenitor cell marker in chromosphere cells and differentiated cells, normalized to GAPDH expression. Displayed are mean values of fold change in Mash1 expression ± standard deviation (n = 3) (∗p < 0.05). Hes5, hairy enhancer of split 5; Sox1, sex-determining region Y box 1; TH, tyrosine hydroxylase; MAP2, microtubule-associated protein 2; Pax6, paired box protein 6; PNMT, phenylethanolamine N-methyltransferase.
Immunostaining for Neural Markers
In addition to the ability to self-renew, stemness is defined as the capacity of progenitor cells to give rise to specific cell types (46,56). To investigate the potential of chromaffin progenitors to differentiate into neurons, chromaffin cells were isolated from eight adrenal glands and cultured with an average yield of 2.25 × 104 cells/gland at day 7 (total amount of 1.8 × 107 cells). After 1 week, chromospheres were enzymatically dissociated, and 7 × 104 single cells were seeded onto cover slips coated with PLL/L and then subjected to differentiation conditions for 8 days. After 1 week, cells gained neural morphology and expressed neural markers, such as β-III-tubulin, TH, and MAP2 (Fig. 4A), as revealed by immunocytochemistry.

Effect of retinoic acid (RA) and/or ascorbic acid (AA) on neural differentiation. (A) Immunofluorescent staining of differentiated chromosphere cells revealed positive staining for TH, β-III-tubulin (left), and MAP2 (right). Scale bar: 50 μm. (B) Quantification of immunocytochemical staining of differentiated chromosphere cells (in presence or absence of RA and/or AA) for β-III-tubulin, TH, and MAP2. Shown are mean percentages of marker positive cells ± standard deviation (n = 3) (∗p < 0.05 and ∗∗∗p < 0.001 vs. standard treatment).
RA and AA Stimulate Differentiation Into Dopaminergic Neurons
Differentiating chromosphere cells were treated with RA and AA, known as potent inducers of dopaminergic differentiation. Indeed, we found a significant (p < 0.001) increase in the differentiation of β-III-tubulin-positive neurons in cells simultaneously treated with 5 μM RA and 10 μM AA (45.3 ± 8.5%) or 10 μM RA and 100 μM AA (44.0 ± 2.9%) as compared to standard treatment conditions. Chromosphere cells that were differentiated in the absence of RA and AA gave rise to 22.9 ± 5.5% β-III-tubulin-positive cells and served as a control (Fig. 4B). A lower but significant increase of β-III-tubulin-positive neurons was induced by incubation with 5 μM RA (37.6 ± 8.3%) or 10 μM AA (31.8 ± 5.7%; p < 0.05) alone, emphasizing a synergistic effect of RA and AA. Unlike neural stem cells (47), a slight increase of MAP2-positive cells emphasized the prevalence of immature β-III-tubulin-positive neurons.
Importantly, dopaminergic neurons increased as determined by immunostaining for TH in parallel to the increase of β-III-tubulin-positive neural phenotype. The frequency of differentiated TH-positive neurons was significantly increased after treatment with 5 μM RA (31.8 ± 9.1%; p < 0.05), 5 μM RA and 10 μM AA (41.6 ± 12.7%; p < 0.05), or 10 μM RA and 100 μM AA (41.8 ± 5.7%; p < 0.001). AA at 10 μM had no significant effect on the number of TH-positive neurons. Chromosphere cells, differentiated without subjecting to RA or AA, were used as a control (21.1 ± 4.2%). Importantly, gene expression of the bicoid-related homeodomain-containing transcription factor (Pitx3) and the orphan nuclear hormone receptor (Nurr1) (Fig. 5A) was significantly increased (p < 0.05) concomitantly with a significant decrease of Mash1 and nestin progenitor markers after RA and AA treatment (p < 0.05) (Fig. 5B).

Fold change in gene expression of nuclear receptor related protein 1 (Nurr1) and paired-like homeodomain 3 (Pitx3) (A), and nestin and Mash1 (B) in the presence or absence (standard treatment) of RA and AA. Displayed are mean values of fold change in gene expression ± standard deviation (n = 3) (∗p < 0.05).
RA and AA Increase Dopamine and Reduce Epinephrine and Norepinephrine Secretion
Functional maturity of neurons derived from chromosphere cells was confirmed by high-performance liquid chromatography (HPLC) analysis of catecholamine release. Neurons derived after incubation with RA and AA were compared to neurons obtained in standard treatment procedure. Catecholamine release from differentiated neurons was measured after 30 min of incubation in extracellular solution with or without 56 mM KCl. Catecholamine concentrations were normalized per 10,000 differentiated cells. Each sample contained 105 differentiated cells per 1 ml. Supernatant from cells treated with RA/AA contained twice the amount of dopamine (7.5 ± 2.6 ng/10,000 cells) as the supernatant of chromaffin cells differentiated by standard protocol (3.5 ± 0.6 ng/10,000 cells; p < 0.05) (Fig. 6A). Enhanced dopamine release was accompanied by a significant increase of GTPCH gene expression (p < 0.05) (Fig. 6B). In parallel, differentiation with 5 μM RA/10 μM AA significantly diminished the release of norepinephrine (24.1 ± 5.2 ng/10,000 cells; p < 0.05) and epinephrine (14.3 ± 9.1 ng/10,000 cells) by approximately 40% compared to standard procedure (epinephrine: 27.7 ± 20.8 ng/10,000 cells and norepinephrine: 44.9 ± 12.7 ng/ 10,000 cells). Moreover, 2.5-fold higher levels of norepinephrine than epinephrine were detected in both standard differentiated cells and cells treated with RA/AA (Fig. 6).

Fold change of catecholamine release from differentiated chromosphere cells and guanosine triphosphate GTP cyclohydrolase (GTPCH) gene expression. (A) Norepinephrine (NE), epinephrine (EPI), and dopamine (DA) concentrations were measured by high-performance liquid chromatography (HPLC). Differentiation with 5 μM RA + 10 μM AA versus standard treatment (basal level line). Catecholamine production was induced by incubation with extracellular solution containing 56 mM KCl (n = 3) (∗p < 0.05). (B) Changes in GTPCH gene expression during standard treatment and differentiation with 5 μM RA + 10 μM AA. Values represent the mean value of fold change ± standard deviation of three independent experiments (∗p < 0.05).
Functional Characterization of Neurons After Treatment with RA and AA
Neuronal function properties gained by differentiation of chromaffin progenitor cells were confirmed by whole-cell voltage-clamping to record voltage-gated sodium and potassium currents after 8 days of differentiation. Activity of neurons differentiated by RA and AA was compared to control cells differentiated by standard procedure. Passive membrane properties of control cells revealed a resting membrane potential of −43 ± 17 mV, an input resistance of 2.7 ± 1.6 GΩ, and the cell membrane capacity of 10 ± 8 pF (n = 12). RA/AA treatment revealed no significant difference: resting membrane potential of −44 ± 20 m V, input resistance of 2.8±1.8 GΩ, and cell membrane capacity of 15 ± 5 pF (n = 15). Seventy percent of differentiated cells using standard treatment (7 of 10 cells) and 60% of cell differentiated in the presence of RA/AA (9 of 15 cells) showed inward currents with voltage dependence and kinetics typical for voltage-activated Na+ channels (Fig. 7A–C). Ninety percent of control cells (9 of 10 cells) and 93% of RA/AA-treated cells (14 of 15 cells) displayed outward current with typical kinetics for delayed rectifier potassium channels (Fig. 7A, B, D). Current clamp recordings revealed repetitive action potential firing in control cells and RA/AA-treated cells, indicating the maturation in fully functional neurons (Fig. 7E, F).

Electrophysiological recordings on neurons derived from chromosphere progenitors. (A, B) Representative voltage-clamp traces from cell differentiated with standard treatment (A) and in the presence of RA + AA (B). For voltage-clamp measurements, cells were held at −80 mV and depolarized in 10 mV steps between −80 and +40 mV. (C) Peak current–voltage relationship for the inward currents with leak subtraction (n = 5; standard error of the mean was around 15% and error bars are omitted in the diagram for clarity). The lines represent fits to the following equation: I(V)/Imax = g(V - Vrev) / (1 + exp[(V - V0.5)/kV]), with I (Imax) being the (maximum) membrane current, g the maximum conductance, V the applied voltage, Vrev the reversal potential for Na+, V0.5 the potential of half-maximal activation, and kV a slope factor. (D) Current–voltage relationship of the outward currents recorded with leak subtraction. (E, F) The firing of repetitive action potentials in response to depolarizing current pulses of increasing amplitude as shown in representative current-clamp recordings from neurons resulted from standard treatment (E) and RA + AA treatment (F).
Discussion
SA progenitor cells from the adrenal medulla bear the potential for cell replacement strategies in neurodegenerative diseases, that is, Parkinson's disease (PD). Our study demonstrates the differentiation of these progenitor cells into functional dopaminergic neurons.
A subpopulation of proliferation-competent SA progenitor cells is presumed to persist in the adult adrenal medulla, based on the fact that chromaffin cell number is regulated to meet physiological requirements. Furthermore, some adrenomedullary tumors seem to develop from SA progenitors within the adrenal medulla; this hypothesis is based on the fact that these pheochromocytomas overexpress multiple genes that are involved in early adrenal and neural development (18,48,54).
We implemented the enrichment of SA progenitors and the differentiation of these progenitor cells using a two-step in vitro protocol. In the first step, the induction of chromosphere formation was achieved in selective proneural culture conditions. Mash1 is a key factor for the development of chromaffin cells expressed in the majority of SA progenitors during embryogenesis (24,31,69). The proneural gene Mash1 is the mammalian homolog of the Drosophila achaete-scute complex encoding the helix–loop–helix type transcription factor achaete-scute complex-like 1 (Acsl1) (33) present in developing brain (41) preceding neural differentiation (37) or contributing to the conversion of human fibroblasts into dopaminergic neurons (53). Upregulation of Mash1 in chromosphere indicates the enrichment of SA progenitor cells in chromosphere in addition to the previously shown increase of the neural progenitor markers Sox1, Sox10, nerve growth factor receptor (NGFR), Musashi1, vimentin, and nestin (10).
Chromospheres, however, are heterogeneous structures consisting of progenitor and chromaffin cells. Along with progenitor markers, the expression of PNMT suggests the presence of differentiated chromaffin cells. Our previous data from single cell RT-PCR indicate that the PNMT-expressing cell population does not overlap with the nestin-expressing progenitor population (10). Expression of the epinephrine synthesizing enzyme PNMT in chromaffin cells is regulated by glucocorticoids (3,11). Therefore, the PNMT expression observed in chromospheres might be due to the glucocorticoids present in the neural stem cell culture supplement (B27). Based on the original work by Wurtman and Axelrod (73), it is generally accepted that glucocorticoids induce the differentiation of chromaffin cells and the expression and function of PNMT. Strikingly, however, SA progenitors are present in chromospheres despite the existence of glucocorticoids in the culture medium, indicating that factors other than glucocorticoids are necessary to induce the differentiation of these progenitors to epinephrineproducing chromaffin cells. This indicates the existence of SA progenitors in the adrenomedullary environment despite the high local glucocorticoid concentrations released from the adrenal cortex.
Progenitor cells in chromospheres self-renew and form clonal secondary spheres (10). To estimate the sphere initiation capacity of chromosphere cells, we now determined the capacity of single cells derived from primary spheres to form secondary spheres within 21 days of culturing. The frequency of formed secondary spheres depends on the density of seeded single cells (71). This method revealed an insignificantly reduced frequency of secondary chromosphere formation after 4 weeks of culturing, indicating slightly reduced proliferative capacity of the cell population selected in neural culture conditions. In addition, to circumvent potential effects that might irreversibly change progenitor properties and impact differentiation capacity or engraftment ability, we decided to investigate chromosphere's differentiation capacity 1 week after isolation. This notion is based on our previous findings where prolonged culturing of neurosphere stem cells resulted in accumulation of chromosomal aberrations and consequent irreversible transformation of stem cell culture (71).
In the second step, cells were subjected to a differentiation protocol consisting of N2 differentiation supplement that promoted the differentiation into a dopaminergic neural phenotype. Under these differentiation conditions, 1-week-old chromosphere cells derived neural cells positive for β-III-tubulin, MAP2, and TH.
These phenotype changes were the outcome of underlying molecular mechanisms that control neural differentiation and functional maturation. During neural differentiation, a simultaneous downregulation of specific neural progenitor markers, such as Mash1, and upregulation of genes specific for differentiated neuronal cells β-III-tubulin, MAP2, and TH were detected. In parallel, the expression of Pax6 was increased; Pax6 is a transcription factor involved in neural differentiation (19,75). Members of the Hes family are downstream effectors of the Notch pathway that play a key role in maintaining neural progenitors in an undifferentiated state (5,27,34). Particularly during early development, the Hes family is engaged when neural progenitors undergo expansion in neural ectoderm (5). In accordance, the expression of neural progenitor markers Notch, Hes1, and Hes5 was reduced during differentiation of chromosphere cells, indicating the same regulatory mechanisms as in the developing nervous system where Hes1 and Hes5 effector expression is correlated with Notch expression (26). In addition, previous studies revealed elevated sphere-forming ability in Hes5-overexpressing multipotent neural stem cells (5). Therefore, these data further suggest that spheroid clusters are supportive structures for the in vitro maintenance of progenitors.
Sox1 is a transcription factor from the SoxB1 subgroup that maintains a stem cell-like state and is a widely used marker of neural stem cells (7). Thus, neurosphere-forming ability was observed in multipotent Sox1-positive cells isolated from cerebellar cortex (1). In accordance, we previously reported an upregulation of Sox1 in chromospheres compared to chromaffin cells (10). Our present data reveal that the shift from spheroid clusters to a differentiated state was accompanied by the downregulation of Sox1 and Sox10, a marker of neural crest derivatives from the SoxE subgroup of transcription factors. The expression of Sox10 seems to be a requirement for undifferentiated neural crest cells to migrate to the adrenal anlagen during development (55).
Interestingly, PNMT expression was diminished regardless of the presence of glucocorticoids due to dominant proneural environmental conditions that instructed SA precursors to launch neural differentiation program. These findings indicate the ability of progenitor cells to derive neural cells despite the presence of pro chromaffin signals in vitro.
The neuronal differentiation capacity was significantly increased by twofold by treating the differentiating cells with RA and AA. Both RA and AA have been shown to launch neural differentiation programs in stem cells and progenitors. RA plays a relevant role during embryogenesis by instructing neural patterning and neural differentiation (42). Furthermore, RA plays an important role in adults by maintaining differentiated state via neural patterning or neural growth. Compared to untreated cells, we observed a significant increase of β-III-tubulin+ and TH+ neurons with no change in MAP2-positive neurons upon treatment with RA and AA. RA and AA had a synergistic effect in inducing neural differentiation. Consequently, RA and AA treatment significantly reduced expression of progenitor markers nestin and Mash1. Importantly, TH increase was accompanied by an upregulation of its activator, the transcription factor Pitx3 (35,58), indicating dopaminergic differentiation of progenitors. In the brain, expression of Pitx3 is exclusively confined to the differentiation of midbrain dopaminergic neurons where it is required for positive control of TH expression (36). Furthermore, recent data demonstrated establishment of Pitx3 as selection marker for dopaminergic neurons derived from embryonic stem cells (78). The orphan nuclear hormone receptor Nurr1 is an important factor in the development of DA neuron progenitors (52). Mice that harbor null alleles of Nurr1 failed to express TH (40,77). Therefore, in these animals, the initiation of catecholamine synthesis and the consequent maturation of dopaminergic neurons failed. Consistently, our findings indicate enhanced dopaminergic neural differentiation since Nurr1 expression was significantly elevated after treatment with RA and AA.
Initially, increased derivation of dopaminergic neurons from neural stem cells after AA treatment was considered as consequence of scavenged oxidative stress (4). Later findings proved the existence of AA-responsive genes that control dopaminergic differentiation (74,76). Our findings could indicate the existence of AA-responsive genes that regulate the differentiation into β-III-tubulin+ neurons from chromaffin progenitors.
Furthermore, we analyzed specific properties of neural function gained after RA and AA treatment. Synthesis of catecholamines was significantly altered after KCL induction and resulted in a twofold increased dopamine production, while norepinephrine and epinephrine production was reduced, indicating the differentiation of chromosphere cells into dopaminergic neurons. Importantly, secreted dopamine levels were similar to the amounts secreted from 21-day cultured mesencephalic neurons (20,57), indicating that differentiated dopaminergic neurons gained crucial functionality important for future transplantation studies. Consistently, elevated dopamine levels were positively correlated with an upregulation of GTPCH expression, the rate-limiting enzyme for dopamine biosynthesis (72). Reduced GTPCH activity leads to the depletion of dopamine and to dystonia in patients affected by Parkinson's disease (51). These findings strongly suggest the gain of dopaminergic function during neural differentiation of chromaffin progenitor cells. The functionality of these differentiated neurons was validated by whole-cell voltage-clamp recordings. The differentiated cells acquired all major functional properties of mature neurons, such as existence of sodium currents and generation of action potentials similar to primary neurons in culture (9,29,44), differentiated neural stem cells from various origins (29,43,59,65), or dopaminergic neurons derived from fibroblasts or neural progenitors (53,65). Similar to most stem or progenitor cell-derived dopaminergic cultures after short-term differentiation periods (8,53), we did not observe spontaneous electrical activity, which usually occurs only after a very long-term differentiation period of 1 month or more (30,64).
The first preliminary data have previously shown that transplants of cultured differentiated neuron-like cells derived from adult chromaffin cells (13,14,49) were able to reduce symptoms in nigrostriatal lesioned rats (13) and a PD patient (14). This proof of principle indicates that the transplantation of differentiated chromaffin progenitor cells can be a future option in the treatment of PD patients.
This study provides evidence for the existence of SA progenitors in adrenal medulla with a pronounced potential to give rise to dopaminergic neurons. The gain of functional characteristics of differentiated neurons raises the possibility to use these cells as valid source of somatic progenitor cells for the treatment of neurodegenerative diseases such as Parkinson's disease. We suggest that the isolation, propagation, and differentiation of chromaffin progenitors from the adult adrenal medulla may be a strategy worth considering for autologous transplantation and cell-based therapy in neurodegenerative diseases.
Footnotes
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft DFG SFB 655 “From cells to tissues” (M.E.B., S.R.B., A.S., and M.A.), DFG KFO 252 “Microenvironment of the Adrenal in Health and Disease” (EH 161/5-1 to M.E.B. and El 855/1-1 to G.E.), and The Center for Regenerative Therapies, Dresden (CRTD). We thank Jochen Haas for technical assistance in adapting the differentiation assay. The authors declare no conflicts of interest.