
Abstract
Hepatocyte transplantation (HT) has become an effective therapy for patients with metabolic inborn errors. We report the clinical outcome of four children with metabolic inborn errors that underwent HT, describing the cell infusion protocol and the metabolic outcome of transplanted patients. Cryopreserved hepatocytes were used as this allows scheduling of treatments. Functional competence (viability, cell attachment, major cytochrome P450 and UDP-glucuronosyltransferase 1A1 activities, and urea synthesis) and microbiological safety of cell batches were assessed prior to clinical use. Four pediatric patients with liver metabolic diseases [ornithine transcarbamylase (OTC) deficiency, Crigler–Najjar (CNI) syndrome, glycogen storage disease Ia (GSD-Ia), and tyrosinemia type I (TYR-I)] underwent HT. Indication for HT was based on severity of disease, deterioration of quality of life, and benefits for the patients, with the ultimate goal to improve their clinical status whenever liver transplantation (LT) was not indicated or to bridge LT. Cells were infused into the portal vein while monitoring portal flow. The protocol included antibiotic prophylaxis and immunosuppressant therapy. After HT, analytical data on the disease were obtained. The OTC-deficient patient showed a sustained decrease in plasma ammonia levels and increased urea production after HT. Further cell infusions could not be administered given a fatal nosocomial fungus sepsis 2 weeks after the last HT. The CNI and GSD-Ia patients improved their clinical status after HT. They displayed reduced serum bilirubin levels (by ca. 50%) and absence of hypoglycaemic episodes, respectively. In both cases, the HT contributed to stabilize their clinical status as LT was not indicated. In the infant with TYR-I, HT stabilized temporarily the biochemical parameters, resulting in the amelioration of his clinical status while diagnosis of the disease was unequivocally confirmed by full gene sequencing. In this patient, HT served as a bridge therapy to LT.
Keywords
Introduction
Children suffering metabolic diseases as a result of liver enzymatic/transport deficits are potential candidates for orthotopic liver transplantation (LT). However, this approach may not always be the first therapeutic option due to the recipient's age, the invasiveness of the surgical procedure, frequent concomitant multiorgan damage, and lack of suitable liver donors (13,16,18). Hepatocyte transplantation (HT) has emerged as a therapeutic strategy, which has been applied with variable success to treat patients for whom LT is not the first clinical indication. Despite HT is presently not being considered a curative therapeutic option, amelioration of patients' clinical status and severity of disease symptoms has been observed in many cases following HT (4,12,19,22,26,27,31,38,43,47,50). The rationale behind hepatocyte cell therapy is based on the hypothesis that transplanted mature hepatocytes, when implanted into the host liver, can undertake metabolic functions that are lacking or badly perform in patients (16,18). Patients with different metabolic abnormalities [i.e., Crigler–Najjar type I (CNI) syndrome, factor VII deficiency, urea cycle defects, glycogen storage disease (GSD) types Ia and Ib, familiar hypercholesterolemia, and Refsum disease, among others] have been treated with satisfactory clinical results (4,10,12,19,22,26,27,29,31,38,47,54).
There is a general agreement that a cellular mass representing a fraction of the whole liver mass (i.e., 5–10%) could be sufficient (depending on the nature of the disease) to restore the function lacking in the damaged liver. However, the number of hepatocytes to be infused each time and the scheduling of cell infusions to treat metabolic disorders reported by clinical researchers vary significantly since distinct metabolic liver diseases may require different degrees of liver repopulation with donor cells to achieve stable correction (1,10,18,19); as yet, no standardized protocols have been established. One additional explanation for such different claims could also be the metabolic performance of infused hepatocytes and their engraftment efficiency into the liver.
To date, no definitive consensus has been reached on the use of either freshly isolated or cryopreserved hepatocytes as well the quality assessment of hepatocytes for HT (11,12,22,26,27,37,46,47,50). Some clinical researchers do prefer the use of freshly isolated cells, as they may retain better metabolic performance than cryopreserved ones. However, to overcome the constraints that limit its wider application, cryopreservation appears the most appropriate form of cell banking, as it allows both programmed and emergency treatments, as in acute liver failure. In this context, refinements of freezing protocols to better preserve hepatocyte functionality have been recently proposed (12,48), which significantly improve the performance of cryopreserved cells after thawing.
In addition, cryopreservation allows a thorough quality assessment and safe characterization of each cell batch prior to its clinical use and seems the only compatible approach for global GMP production of cells. By adopting a strategy consisting of the cryopreservation of only highly viable hepatocytes (>80%), improved freezing protocols and routinely assessing their metabolic functionality and microbiological safety after thawing and before clinical use, selection of good metabolic performing, and safe hepatocyte batches could be ensured prior to each HT (8,13,14).
This work reports the clinical outcome of four pediatric patients suffering different metabolic diseases (ornithine transcarbamylase [OTC] deficiency, Crigler–Najjar [CNI] syndrome, glycogen storage disease Ia [GSD-Ia] and tyrosinemia type I [TYR-1]) who underwent hepatocyte transplantation with quality-assessed cryopreserved human hepatocytes to ultimately improve their clinical status and to eventually avoid, postpone, or bridge whole-liver transplantation.
Materials and Methods
Hepatocyte Isolation From Human Liver
The cell isolation process was totally conducted in a clean room (ISO class 6) in accordance with European guidelines EN-ISO-14644 (8,14). Human hepatocytes were prepared from livers that were procured but subsequently rejected for LT for different reasons (i.e., prolonged cold ischemic time, mild fibrosis, arteriosclerosis, neonatal donors, damaged arteries, blood vessels, or capsules) under the supervision of the hospital's transplantation coordination and ethics committee. These livers were free of hepatitis viruses and human immunodeficiency virus (HIV) and were donated by relatives for research use according to legal protocols. Once extracted from the cadaveric donor, liver tissue was flushed and maintained in ice-cold Celsior solution until cell isolation (41). Cold ischemia never exceeded 8 h (Table 1). The liver tissue was enzymatically dissociated by the two-step collagenase perfusion technique, as described in detail elsewhere (8,14,41). Once isolated, cells were purified by sequential centrifugation and washed in an appropriate medium, as described in detail (8,14). Hepatocyte viability was assessed by the dye exclusion test with 0.4% Trypan Blue. Only suspensions with viabilities greater than 80% underwent further characterization and cryopreservation.
Details of Donors, Organs, and Isolation of Hepatocytes

F, female; M, male.
Mandatory environmental clean room monitoring was routinely carried out, including microbiological testing of personnel, air, surfaces, and media samples. Microbiological controls were also run in all the steps of cell isolation, cryopreservation, and thawing. Records of all the analyses were independently monitored.
Hepatocyte Cryopreservation and Thawing
Freshly isolated hepatocytes were pelleted and thereafter gently dispersed in ice-cold University of Wisconsin solution containing 10% dimethyl sulfoxide. They were distributed in 50-ml freezing bags, each containing 20×106 cells/ml. Cells were cooled using a controlledrate freezer and were finally stored in liquid nitrogen at −196°C until use, as described in detail elsewhere (14). Twin vials containing cryopreserved cells were used as a regulatory safety requirement for quarantining cells against bacterial or fungal contamination, as well as to monitor the metabolic and functional performance of cells after storage and thawing. Bacteriological controls were run for samples taken at all stages of cell isolation, cryopreservation, and thawing.
Just before conducting HT, the requested frozen bags were placed into a water bath at 37°C and gently stirred. Thawed cells were washed once with 10 volumes of infusion media at 4°C, centrifuged at 50xg for 5 min, and were finally resuspended in the infusion medium prior to use (21).
Functional Assessment of Banked Hepatocytes
Each batch of frozen hepatocytes was routinely tested prior to clinical use. Upon thawing an aliquot of frozen cells, viability and attachment to collagen/fibronectin-coated plates were assayed (14). Drug-metabolizing activities were also monitored by incubating thawed hepatocytes in suspension with a cocktail of appropriate cytochrome P450 (CYP) substrates. The following activities were measured: 7-ethoxyresorufin O deethylation (CYP1A2), coumarin 7-hydroxylation (CYP2A6), diclofenac 4′-hydroxylation (CYP2C9), chlorzoxazone 6-hydroxylation (CYP2E1), and midazolam 1′-hydroxylation (CYP3A4) (8,14). Conjugation of β-estradiol with glucuronide by phase II enzyme uridine diphosphate (UDP)-glucuronosyltransferase 1A1 (UGT1A1) was also determined (8). The enzymatic activities present in the hepatocytes were quantified by measuring the formation of the corresponding metabolite by HPLC/MS/MS, according to previously published methods (25). Ureogenesis was assessed in the thawed cells by measuring the formation of urea from NH+4, as described (14).
Patients
Four pediatric patients with inborn errors of liver metabolism underwent HT. The clinical HT protocol was approved by the hospital's ethical committee and by the transplants committee. Informed consent was obtained from parents prior to HT, which was conducted under the premises of the 1975 Declaration of Helsinki and subsequent amendments.
Complete blood cell count, biochemistry (glucose, ammonia, blood urea, creatinine, total proteins, serum electrolytes, uric acid, triglycerides, lactic acid), liver function parameters (glutamic oxaloacetic transaminase [GOT], glutamic pyruvate transaminase [GPT], alkaline phosphatase [AP], total bilirubin, direct and indirect, cholesterol, albumin, prothrombin time [international normalized ratio (INR)]), and specific metabolic parameters according to the child's metabolic disorder were obtained upon admission to hospital.
The diagnoses of each metabolic disease were unequivocally established on the basis of liver enzymatic activities measurement and/or specific serum metabolites and were confirmed by genetic studies. Each patient underwent a complete clinical history and physical examination. Hepatic ultrasonography and portal flow Doppler examination were routinely performed to exclude vascular disorders.
Indication for HT in patients was based on (a) severity and prognosis of the disease, (b) impaired quality of life, and (c) expected benefits, whenever LT was not a therapeutic indication or it had to be postponed. In all four patients, the therapeutic measures previously taken had failed to stabilize the disease, there were clear analytical and clinical data evidencing clinical deterioration, and no contraindication for HT was initially found. The patients stayed in the Paediatric Intensive Care Unit (PICU) for at least 24 h for observation purposes. Upon admission to the PICU and for monitoring after HT, blood and urinary tests were routinely performed every 12 h. Thereafter, the clinical response was periodically monitored by laboratory data (including the relevant clinical parameters for each metabolic disorder) and by clinical scores.
Portal Venous Access
Catheterization of the inferior mesenteric vein was followed by placing a silicone catheter (port-a-cath®) with a size from 4.5 to 7.0 french (depending on the patient's weight) into the portal vein, connected to a port device positioned subcutaneously on the left thoracic cage. This was performed under general anesthesia by open surgery or laparoscopic access. Before portal vein catheterization, fresh frozen plasma and/or platelets administration was performed when indicated; after placement and after each hepatocyte infusion, the port-a-cath device underwent routine preventive heparinization (dose, 100 UI/kg/day). On discharge, catheter supervision and care were carried out monthly. This permanent and long-lasting access allowed repetitive intraportal infusions and helped outpatients lead a normal life.
Hepatocyte Infusion
Hepatocytes were selected from ABO compatible donors and infused via the portal vein. Infusions were performed in either the PICU or the Pediatric Transplant Unit under continuous cardiovascular monitoring and mild sedation (benzodiazepines) conditions whenever necessary. Preventive hospitalization after each successive hepatocyte infusion was ensured for 48–72 h. The clinical response was periodically monitored by laboratory data (the relevant clinical parameters for each metabolic disorder) and clinical scores. Before cell infusions, patients received prophylactic treatment with antibiotics.
As part of the immunosuppression protocol, an intravenous bolus of hydrocortisone was administered 1 h before hepatocyte infusion at a dosage of 10 mg/kg, followed by methylprednisolone 1 mg/kg/day in between infusions, plus tacrolimus 0.1 mg/kg/day. Trough blood levels of tacrolimus were monitored every 24–48 h and dose-adjusted to reach concentration ranges in between 5 and 10 ng/ml. After HT, steroids were progressively tapered.
The number of cells we initially intended to infuse into our patients was between 5% and 10% of the patient's theoretical liver mass distributed in the several infusions administered, assuming that liver contains about 1.8 × 108 hepatocytes/g of tissue (18,40).
Hepatocytes were suspended in an appropriate volume of infusion medium (ca. 30–50 ml) at a density ranging between ca. 10 and 15 × 106 hepatocytes/ml (13,21) and were infused manually at a flow rate of 1 ml/min, with frequent and gentle waving to keep cells in suspension (13,42). The infusion was performed for 40–60 min under strict sterile conditions. Cells were administered to patients in repeated cell infusions at given time intervals, taking into account the recommendations given by other authors concerning the number of cells per gram of liver that can be safely transplanted in a single procedure (30–100×106/kg of body weight or 2.4×106 cells/g of liver) (13,17,18,40).
Color Doppler ultrasound imaging (CDU), prior to, at 15-min intervals, and at the end of each infusion, was performed to control catheter position and to monitor portal flow (19,30,33). Further ultrasound monitoring was scheduled at 12, 24, and 48 h post-cell infusion. Depending on the patient's clinical course after HT, new cell infusions were scheduled and administered.
Results
Viability and Functional Assessment of Hepatocytes
The human livers used as source for hepatocytes for clinical use had cold-time ischemia times that did not exceed 8 h. Only one donor was in asystole for 50 min (warm ischemia). Cell yield and viability were monitored with each hepatocyte batch preparation (Table 1). Under the standardized conditions used, cell viability of freshly isolated hepatocytes was consistently greater than 80% (average value, 86±6%), with an average yield of 6.5±1.2×106 cells/g of tissue. Only those suspensions with viabilities greater than 80% underwent cryopreservation. Upon storage at −196°C, cell viability, attachment, and metabolic functionality were periodically assessed by thawing a vial of each batch of cryopreserved hepatocytes (Table 2). The average cell viability of the cell batches used in this clinical study after thawing was 66±7% (Table 2). The attachment efficiency was also monitored (cell attachment to collagen/fibronectin-coated plates substrate 1 h after cell plating) and was 45±13% (Table 2). In our hands, 10–20% of these initially attached hepatocytes underwent apoptosis in the 24 h following cell plating (data not shown).
Functional Characterization of Cryopreserved Hepatocytes

The ureogenic rate was expressed as nanomol per minute per million of cells, and the P450 activities were expressed as picomol per minute per million of cells. CYP, cytochrome P450; EROD, ethoxyresorufin O-deethylation; C7OH, coumarin 7-hydroxylation; D4OH, diclofenac 4′-hydroxylation; C6OH, chlorzoxazone 6-hydroxylation; M1OH, midazolam 1-hydroxylation; UGT1A1, uridine diphosphate (UDP)-glucuronosyltransferase 1A1; β-EST-G, β-estradiol 3-glucuronidation.
Cryopreserved/thawed hepatocytes' biotransformation competence was routinely assessed by measuring the activity of major human CYPs and the UGT1A1 conjugating enzyme. The results displayed in Table 2 indicate that the hepatocytes used in cell transplantation all met the criteria of expressing phase I and II activities comparable to those of freshly isolated hepatocytes from normal livers (20). Cryopreserved/thawed hepatocytes were able to synthesize urea from ammonia at good rates (0.76–3.84 nmol/min/106 viable cells) (Table 2).
Clinical Features of Transplanted Patients
Four patients suffering from liver-related metabolic diseases were included in this clinical study; Patient 1 (P1) had ornithine transcarbamylase (OTC) deficiency; Patient 2 (P2) showed a Crigler–Najjar (CNI) syndrome; Patient 3 (P3) was diagnosed of a glycogen storage disease type Ia (GSD-Ia); Patient 4 (P4) had tyrosinemia type I (TYR-I). Patients had previously been diagnosed and treated accordingly. HT was only considered when previously undertaken therapeutic measures in these patients according lex artis had failed to stabilize their diseases and there were analytical and clinical data evidencing a progressive clinical deterioration.
During the preparatory phase (placement of the port-a-cath® device), patients did not show any specific complications. The cell infusion stage was well tolerated by all patients without causing them noticeable discomfort. There were no severe and/or irreversible vascular complications during or after HT in any patient treated. During infusion, portal blood was continuously monitored (Fig. 1). Patients 1 (P1), 2 (P2), and 3 (P3) showed no noticeable alterations, and their portal blood flow remained constant and normal at all times, both during and after cell infusions. Patient 4′s (P4) portal flow transiently decreased after cell infusion but recovered within the following hours (Fig. 1); conditions were modified in the subsequent cell infusion to minimise this effect. Sedation was not needed, except for the CNI patient for whom benzodiazepines were used as sleep inducers due to her restlessness.

Changes in portal blood flow upon patients' hepatocyte infusions. The two cell infusions administered to patients 1 (P1) and P4 and the first two infusions administered to P2 and P3 are displayed. Portal flow preinfusion, immediately postinfusion, 12 h postinfusion, 24 h postinfusion, and 48 h postinfusion are shown. HT, hepatocyte transplantation.
Patient 1: Urea Cycle Disorder
P1 was a 12-year-old girl suffering a urea cycle disorder (OTC deficiency) and was diagnosed at the age of 4. The patient had frequent vomiting, decreased level of consciousness, and elevated hyperammonemia (350 μmol/L). Disease diagnosis was confirmed by a genetic study, which evidenced that she carried the c.533 C >T/p.Thr178Met mutation.
A vegetarian diet was initially established, together with pharmacological treatment (sodium phenilbutyrate, arginine, B12 vitamin, and folate). This treatment achieved the patient's clinical stability with minor metabolic decompensation in the following years. However, at the age of 10, her clinical situation dramatically worsened. She went into a hyperammonemic coma and suffered brain edema requiring decompressive craniectomy. The patient partially recovered from this event but showed important neurological aftermath, such as loss of speaking abilities and increasing walking difficulties. Since this episode, she experienced recurrent metabolic decompensation, which led her to continuous and repeated hospital admissions (1–2 per month in the following 8 months). Moreover, her clinical and neurological status severely and progressively deteriorated. In view of her overall clinical situation, the patient was not considered for LT. Following a new episode of acute decompensation, she was referred to our hospital and was admitted to the PICU, where she stayed throughout her hospitalization.
When admitted to hospital, therapeutic measures such as protein restriction (1.5 g/kg/day) and sodium phenylbutyrate (dosage 13 g/m2) were applied. These measures, identical to those adopted in previous decompensation episodes, did not elicit comparable benefits for the patient in terms of biochemical improvement, while her clinical situation worsened.
In view of her unstable condition, HT was considered as a mean to attain a clinically and metabolic rescue, which could eventually be further maintained by conventional medical treatment. The patient initially received 0.87 × 109 hepatocytes (cell batch H4/08), representing 2% of estimated liver mass, administered in two hepatocyte infusions into the portal vein in a 1-week interval. After the first infusion (day 1), the ammonium and glutamine levels initially decreased. However, an intercurrent infective episode resulted in transient metabolic decompensation by day 3, with increased blood ammonia and glutamine; once controlled, the levels of ammonia and glutamine diminished, while that of urea increased over the values recorded prior to HT (Fig. 2).

Evolution of ammonium, glutamine, and urea levels after hepatocyte infusions in the urea cycle disorder (OTC) patient. Arrows show the two hepatocyte infusions on days 0 and 7. An intercurrent infective episode that occurred days later resulted in severe metabolic decompensation on the following 3 days, with increased blood ammonia levels, which were clinically well tolerated, is indicated with an asterisk.
A good response to antibiotic therapy (in absence of immunosuppression treatment) allowed patient recovery and a second hepatocyte infusion (day 8); again, this was followed by metabolic improvement with blood ammonia decreasing by 60% in relation to initial values. The glutamine serum values substantially lowered while urea increased, and this situation persisted over the following days (Fig. 2).
The patient's biochemical parameters substantially improved in comparison with previous hospitalization episodes, despite the small amount of cells infused (Fig. 2). Protein restriction and conventional pharmacotherapy were maintained at all times. Two weeks after HT, while the metabolic improvement was still evident, the patient developed nosocomial sepsis by Aspergillus fumigatus, which was unrelated to cell preparation, along with a poor response to medical treatment despite immediate discontinuation of immunosuppression treatment. Her status rapidly worsened, and the patient died from septic shock 30 days after. Liver cell engraftment could not be assessed as her parents did not authorize an autopsy.
Patient 2: Crigler–Najjar Type 1 Syndrome
P2 was a 7-month-old girl who had been diagnosed with CNI syndrome in the newborn period. Genetic confirmation of the disease was accomplished by completely sequencing the UGT1A1 gene. A nucleotide change c.120 delG (p.Trp40CysfsX2) in homozygosis was detected. Both parents were consanguineous and carried the same mutation. Despite her early diagnosis, she had not received phototherapy treatment from the age of 2–4 months due to her temporary residence in an underdeveloped country. She was referred to our hospital given her high, sustained unconjugated hyperbilirrubinemia.
A physical examination of the patient upon admission evidenced severe jaundice and clinical signs of neurological affection, such as spasticity in both upper and lower extremities, predominantly on the left side with axial hypotonia. A serum analysis showed very high total bilirubin values (25 mg/dl), mostly in the unconjugated form (24.2 mg/dl), with otherwise normal liver function tests. The brain magnetic resonance image (MRI) was compatible with kernicterus, with increased intensity in pale globes. Visual- and hearing-evoked potentials and the electroencephalogram were normal. Daily phototherapy treatment and administration of phenobarbital as an enzyme inducer was established, together with an intensive rehabilitation programme. However, 2 months later, no clear improvement in bilirubin levels was observed.
After assessing the child's neurological impairment and the risk versus benefit of LT, it was decided to postpone her inclusion as a candidate for LT. HT was proposed as an alternative therapeutic approach with the aim of diminishing the need for phototherapy, thus allowing compliance with a more intensive rehabilitation program and a potential improvement of her neurological condition.
The functional characteristics of the hepatocytes used in this patient were among those displaying high UGT1A1 activity (Table 2); the infusion protocol is shown in Figure 3. The total number of transplanted cells was over 6.7 × 109 hepatocytes (17% of liver mass) during a 16.6-month treatment period. The initial treatment consisted in the infusion of 2.66 × 109 hepatocytes in seven consecutive infusions over 1.5 months (7.6% of liver mass) (Table 3 and Fig. 3). Following this cell therapy, a consistent decrease in bilirubin levels down to 14.1 mg/dl was observed. At this point, infusions were discontinued, and the patient underwent a close clinical and biochemical follow-up (Fig. 3). By day 126 postinitiation of the cell treatment, as a weak increase in bilirubin was noted, two new cell infusions (days 126 and 130; 0.6 × 109 hepatocytes) were preventively administered (Fig. 3). Another single cell infusion of 0.5 × 109 hepatocytes was carried at day 203; in both cases, a subsequent tendency to decrease bilirubin levels was noted. By days 269 and 270 postinitiation of hepatocyte therapy, two hepatocyte infusions (0.965 × 109 hepatocytes) were administered with the aim of further decreasing unconjugated bilirubin levels (Fig. 3). By day 485, as a slight increase in bilirubin had been observed, the patient received three additional cell infusions (1.62 × 109 hepatocytes). The transplanted cells again elicited a noticeable decrease in bilirubin levels (Fig. 3).

Changes in the bilirubin levels of the Crigler–Najjar (CNI) patient after hepatocyte infusions. Arrows indicate the timing of each infusion.
Crigler–Najjar Type I (CNI) Patient, Protocol of Infusions, Functional Characteristics of Infused Hepatocytes

Br, bilirubin; GOT, glutamic–oxaloacetic transaminase; GPT, glutamic–pyruvic transaminase; IU, international unit.
In this patient, HT resulted in a decrease and a long-term stabilization of bilirubin levels around 50% of the initial values prior to cell infusion (Fig. 3). In addition to this improvement in the biochemical parameters following HT, the patient's neurological deterioration not only stopped but even her motor skills significantly improved with lessened spasticity. In a recent brain MRI, performed at the age of 2 years, an improvement of the T2 signal in pale globes was observed when compared to the previous MRI taken at the age of 10 months. While prior to HT continuous, phototherapy (24 h) was necessary; after HT, the need for phototherapy decreased to 12 h a day, so it could be discontinued during the day, allowing the patient to attend a rehabilitation center as well as to be taken out by the parents for a daily walk or other activities.
In the past year, the patient has shown stable bilirubin levels without any further HT treatment.
Patient 3: Glycogen Storage Type 1a Disease
P3 was a 6-year-old girl diagnosed with GSD-Ia who was referred to the Unit because of repeated metabolic decompensations (hypoglycemia) with loss of consciousness and important biochemical alterations (hyperuricemia, hypertriglyceridemia, lactic acidosis), which resulted in frequent hospital admissions. Confirmation of the diagnosis was accomplished by genetic analysis, which revealed the mutation p.Arg83Cys in homozygous state in exon 2 of gene glucose-6-phosphatase (G6PC) (i.e., glycogenosis type Ia).
Despite dietary management that included nocturnal nasogastric feeding and low lactose and fructose meals with added uncooked cornstarch (UCCS) every 3 h, she was unstable and unable to maintain blood glucose constantly above 70–105 mg/dl, which is required for a good metabolic control of the disease; in fact, she had frequent hypoglycemic episodes, preventing her from receiving regular schooling. Her levels of lactic acid, cholesterol, and triglycerides remained high, while liver function tests were normal. α-Fetoprotein was within the normal range (2.2 ng/ml). No psychomotor impairment was detected. Ultrasonography showed the presence of two solid lesions in the right liver lobe suggestive of adenomas. However, the MRI excluded this possibility and confirmed the presence of two hemangiomas located at segment V (7 mm) and segment II (5 mm).
HT was considered in view of the patient's clinical instability, frequent hypoglycemic episodes, the patient's age, the GSD-Ia prognosis, and absence of hepatic adenomas. The total number of infused cells was 2.35 × 109 cells (5.97% of estimated liver mass) administered in seven cell infusions over 65 days. Initially, the patient received 0.8 × 109 hepatocytes (cell batch H13/08) in two infusions with a 2-day interval. After this initial treatment, a significant, but limited, improvement in the patient's biochemical parameters was evidenced (Fig. 4). This was followed by a second series of hepatocyte infusions, which were administered 13 days after the previous one. The patient received four infusions along 1 month (0.9 × 109 hepatocytes, batch H1/09), and the biochemical parameters were monitored (Fig. 4). Following a reevaluation of her clinical status and the risks/benefits assessment of treatment, another hepatocyte infusion was scheduled (0.63 × 109 hepatocytes; cell batch H3/09) 1 month later, thus reaching a number of cells equivalent to 6% of the estimated whole liver mass (Fig. 4).
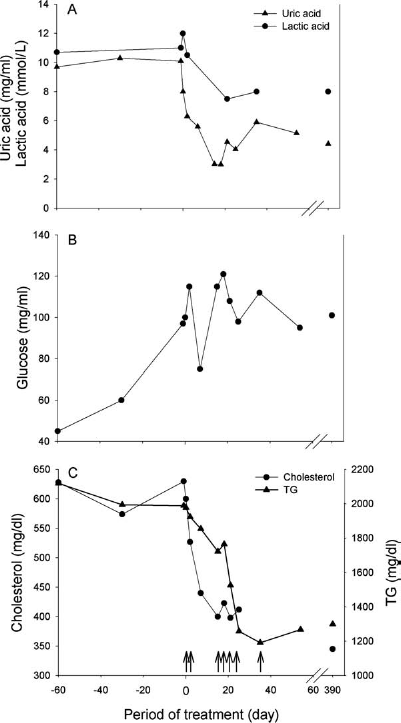
Evolution of biochemical parameters of the glycogen storage disease type Ia (GSD-Ia) patient after hepatocyte infusions. Changes in the levels of uric and lactic acids (A), glucose (B), and cholesterol and triglycerides (TG) (C). Arrows indicate the timing of each infusion.
After the first series of hepatocyte infusions, the patient did not suffer the previously frequent episodes of hypoglycemia and her blood glucose levels and biochemical parameters (lactic acid) significantly improved, although they did not reach normal values (Fig. 4). Complementarily to cellular treatment, the previously established dietary measures were maintained with nocturnal nasogastric feeding, low fructose and lactose meals every 3 h plus UCCS. In between cell infusions, the patient was discharged, and no symptomatic hypoglycemia episodes were observed at home. Even so, regular monitoring of glucose levels showed capillary blood glucose to be around 70 mg/dl before meals and around 120 mg/dl after meals. Morning-fasting glucose levels were between 55 and 72 mg/dl, although only very sporadically was glucose level lower than 60 mg/dl. One year after the last hepatocyte infusion, similar glucose levels were observed, the patient had no symptomatic hypoglycemia episodes at home and did not require hospital admission. As for lactate, levels before referral were in between 11 and 15 mmol/L, and these levels were never so high after HT; most values were usually around 7 mmol/L. She is growing well and is attending school regularly.
Patient 4: Tyrosinemia Type 1
P4 was a 45-day-old male baby who was referred to the Unit because of hypocoagulability (INR 4.7; Quick Index 12%; factor V 23.2%; factor VII 3.2%) and with no other biochemical signs of liver dysfunction (GOT: 26 UI/L, GPT: 12 UI/L, total proteins: 6.2 g/dl, albumin: 3.3 g/dl, fibrinogen; 190 mg/dl). He had been previously admitted to another hospital where a metabolic disease was suspected.
Upon admission, phenylalanine and tyrosine were high in urine, and blood amino acid levels were all high and above range, suggesting nonspecific liver dysfunction. No structural alterations in the liver parenchyma were detected neither by CDU nor MRI upon admission. The repeated liver ultrasound indicated normal portal blood flow and no ascites. A histological study of the liver showed mild nonspecific incipient fibrosis, but no evidence of cirrhosis.
TYR-I was clinically suspected, and a diet poor in phenylalanine and tyrosine and treatment with Nitisinone (NTBC, 1.5 mg/kg/day) were started. Fourteen days of dietary and medical treatment did not show an improvement, and although the patient remained in good condition with no bleeding episodes, bilirubin levels tended to increase and blood coagulation worsened with a progressive decrease in clotting factors V and VII, despite intravenous administration of vitamin K (Fig. 5). The clinical diagnosis of P4 remained unsure because of lack of succinylacetone in urine, despite repeated determinations. Genetic analysis, comprising full sequencing of the fumarylacetoacetate hydrolase gene, was requested.

Evolution of clotting factors (V and VII) and bilirubin levels after hepatocyte infusions and after LT in the tyrosinemia type 1 (TYR-1) patient. Thin arrows show the two hepatocyte infusions on days 0 and 2. The thick arrow indicates the day of whole-liver transplantation (LT) 45 days after the first hepatocyte infusion.
In these circumstances, HT was considered to be a good option to improve the clinical status of the patient, to eventually achieve a late response to NTBC (some patients show a delayed pharmacological response), as well as to obtain the genetic evidences for an unequivocal confirmation of tyrosinemia 1. Forty-eight hours prior to HT, 50 ml of fresh frozen plasma were infused, followed by placing a 4.5-french silicone catheter (port-a-cath®) into the portal vein, to minimize bleeding risks. The baby received 0.66 × 109 hepatocytes (cell batch H3/09) representing 3.5% of estimated total liver mass within 48 h in two infusions. Before and during the first hepatocyte infusion, the CDU revealed normal portal flow. However, a drop in portal flow was detected 24 h after (Fig. 1), followed by complete recovery by 48 h with no clinical symptoms. In view of this, the infusion rate in the second cell administration was lowered (40 ml/h vs. 60 ml/h). Portal flow during the second hepatocyte infusion was normal, but a transient decrease in portal flow was noticed after HT, which was satisfactorily handled by gently increasing the saline solution infusion rate through the port-a-cath® from 5 to 8 ml/h. Portal blood flow started to improve 4 h later with almost complete recovery of the preinfusion values in the following 8 h (12 h after cell infusion), and normalization by 24 h post-HT without clinical consequences (Fig. 1). The subsequent controls showed the catheter to be in place and a normal portal blood flow.
Following hepatocyte treatment, while maintaining the same diet and NTBC treatment, an improvement in clotting factors levels was observed for the first time since admission (Fig. 5), in parallel, bilirubin levels clearly decreased (Fig. 5). After HT, no more fresh frozen plasma was administered.
The patient maintained a stable analytical and clinical status until day 14 after HT when a bacterial infection was detected, which resulted in a transient decrease in factor V levels and a peak in the bilirubin levels (Fig. 5). He recovered with antibiotics and immunosuppressive treatment removal. A CDU performed at this time revealed incipient ascites, while portal blood flow was normal. The portal catheter was removed, but this had no influence on ascites progression. No further cell treatment was attempted, and on day 24, after the first cell infusion, bilirubin started to increase again (Fig. 5).
Meanwhile, tyrosinemia type 1 was fully confirmed after an extensive genetic study, which determined that the patient carried the IVS6-1 G>T and IVS12+5G>A mutations in the fumarylacetoacetate hydrolase gene, both of which relate to bad prognosis. Immediately, the patient was included on the waiting list for cadaveric LT as no suitable candidate for living LT was available. The molecular diagnosis was reconfirmed by a second, independent, genetic laboratory. A cadaveric liver could be obtained within the following 48 h, and the patient was transplanted. Two days after LT, clotting factors reached normal levels, and the patient was discharged 4 weeks later in good clinical condition. Follow-up in the outpatient's clinic was ensured without recording any major clinical event. An analysis of the explanted liver revealed incipient liver cirrhosis.
Discussion
HT could be an option to improve the outcome of metabolic diseased patients who are unstable but not sick enough for LT and for those whose risks associated with LT may not be justified (4,10,12,19,22,26,27,29,31,38,47,48,51). The clinical pros and cons of HT are currently a matter of discussion. The clinical efficacy is likely to rely on many different factors, among them, the nature of the disease, the number of cells required/administered, their functional performance (i.e., ability of transplanted hepatocytes to assume the metabolic functions lacking in the diseased liver), and ability of cells to engraft and survive in the host organ (8,13,14,16,18,21,25). As compared to LT, HT is less invasive, seems well tolerated by patients, and does not contraindicate or preclude LT overtime. The present paper reports our experience in HT performed in four paediatric patients affected by inborn metabolic diseases in which lacking enzyme activity is primarily expressed in the liver using well-characterized cryopreserved human hepatocytes.
To date, no general consensus on the use of either freshly (12,22,46,51) isolated or cryopreserved hepatocytes for HT has been reached (12,26,27,47). Although freshly isolated cells better retain their functionality (45,48,52,53), there are several reasons in favor of using cryopreserved hepatocytes for HT. Firstly, it better complies with regulatory safety requirements, allowing effective quarantining of cells against bacterial or fungal contamination prior to their use. Secondly, it facilitates prescheduled and repeated infusions to patients. Thirdly, it allows the assessment of the metabolic activities of banked hepatocytes in helping make decisions about the quality and suitability of a given hepatocyte batch for a given clinical use/patient (8,14,23) since the outcome of HT is closely related to the quality of the transplanted hepatocytes (37).
A cellular mass representing 5–10% of whole liver mass administered in repeated cell infusions is claimed by clinical researchers as being capable to restore the metabolic liver function, which is missing in inherited metabolic-diseased patients. In spite of there being no formal consensus, most authors (2,13,19,31) suggest that the mass of cells required to correct a single enzymatic defect is likely to be less than for the treatment of chronic disease or acute liver failure as claimed by others (42,54). Indeed, it has been reported from 4% to 8% of total liver mass in CNI patients to just 1% of total hepatocyte mass in a patient GSD-Ia, to achieve clinical improvement (31). In addition to the disease and patient's clinical status, this figure is likely to be related to the functional status of transplanted hepatocytes (2,12,19,26,36,42,47). In our study, we transplanted between 2% (OTC patient) and 17% (CNI patient after 16.6 months) of total liver mass in repeated infusions in relation to the patients' clinical evolution. However, the mass of cells that can be safely infused each time into the liver is restricted by the portal hypertension effect, and consequently, this limits the number of hepatocytes to be transplanted and the number of total infusions (4,10–13,19,22,26,27,30–32,38,47,48,51). Some authors indicate that no more than 1–2% of liver mass can be transplanted in each single cell infusion (18,40).
The indication for HT in children is a matter of discussion. In our case, patients underwent this treatment only when all the therapeutic measures taken had failed and LT could be delayed or avoided if the patient's status improved (4,10,12,19,22,27,29,38,47). For P1, LT was excluded due to her neurological status; for P2 and P3, LT was not an urgent need and it could be postponed if HT improved the metabolic status of the patient. In P4, we were confronted with slight clinical deterioration of the patient, while conclusive diagnosis was still not available; in this case, HT was considered solely as a bridge to LT, and once the genetic diagnosis was fully confirmed and chance to obtain any response to the pharmacological treatment was very low, the patient was transplanted.
The site and route of hepatocyte infusion are relevant. In this clinical study, hepatocytes were infused into the portal vein through a catheter implanted via the inferior mesenteric vein and connected to a port device (port-a-cath®) positioned subcutaneously on the left thoracic cage to facilitate subsequent cell infusions. The route and site have been previously acknowledged by researchers as being the most successful way to transplant hepatocytes into children with inborn errors of metabolism (4,12,19,22,27,47). This route is preferred when there is no indication of liver flow alteration. In adults, where the most frequent indication for HT has been acute liver failure, frequently associated with liver cirrhosis, hepatocytes are frequently infused into the splenic artery (7,50,51).
The complications of HT so far reported include transient portal hypertension, portal flow reduction, transaminases elevation, and portal vein thrombosis (5,19,31,51). Data on portal vein pressure after HT are available in the scientific literature for 18 patients so far. Portal vein pressure temporarily increased during cell application in five of six adults (31,39) and in 11 of 12 children (4,12,19,22,38,43,51). In general, these effects are transient. Additionally, Strom et al. reported decreased portal blood flow after infusing hepatocytes into a patient with α1 antitrypsin deficiency that recovered within the following 24–48 h with no clinical signs (54). As in other cases reported in the literature, in our patients, portal vein pressure was indirectly monitored through portal flow measurement by Doppler ultrasound in order to keep a very narrow control on portal pressure while minimizing catheter manipulation (46). In three of the four treated patients, no alteration of portal blood flow was observed, but a transient reduction was noted in P4 diagnosed with TYR-1, 24 h after the first infusion, that recovered normal values within the following 24 h (Fig. 1). Consequently, we lowered the rate of perfusion of the second hepatocyte infusion to 65% of that used the first time, and although a decrease in portal flow was again detected, it was properly managed and the recovery of portal blood flow to preinfusion values was reached within the following 8 h and without clinical symptoms.
The first patient to receive HT was a 5-year-old boy with OTC deficiency in 1997 (51). To date, HT has been reported in the literature in nine patients with urea cycle defects: six with ornithine transcarbamylase (OTC) deficiency, one with carbamoyl phosphate synthetase (CPS1) deficiency, one with citrullinemia, and one with adenylosuccinate lyase (ASL) deficiency (22,29,38,47,48,51). HT resulted in these patients in a variable degree of metabolic stabilisation, with decreased plasma ammonia levels, and increased urea production and protein tolerance (22,38,46,47). Our patient had been diagnosed at the age of 4 years and reasonably managed until the age of 10 years until her clinical situation dramatically worsened. Since this episode, she experienced recurrent metabolic decompensation and repeated hospital admissions, and previous clinical management by dietary protein restriction and pharmacological treatment failed to control and stabilize the disease, resulting in severe neurological impairment. Our patient had no indication for LT because of her advanced state of metabolic and neurological deterioration. HT has offered her to improve her quality of life with the aim of diminishing the frequency of decompensation episodes and continuous hospital admissions.
We were aware of the potential risk of sepsis by introducing a port catheter and starting immunosuppression, but not many alternatives were at hand considering the high number and severity of decompensations the patient had suffered in the months prior to this last one.
After HT, blood ammonia progressively declined, together with a substantial decrease in glutamine values; this was paralleled by an increase of urea production, indicating that a significant number of hepatocytes must be metabolically active after transplantation. Only two infusions could be applied as her clinical status became complicated by a nosocomial fungal sepsis unrelated to HT, and the treatment was discontinued. However, the metabolic effects observed (a reduction of almost one third of the initial ammonia levels) reveal that unequivocal metabolic changes can be obtained shortly after an infusion of a relatively low cell mass; this is in agreement with what others have reported (22,47). Of the six patients with OTC deficiency worldwide that underwent HT so far, none have resulted in a resolutive treatment; they were transplanted thereafter or entered in the waiting list for LT (22,29,38,47).
In CNI disease, the liver parenchyma architecture and metabolic functions are normal, except for UGT1A1 activity. The patient is at risk of severe neurological complications given the inability to conjugate bilirubin because of the lack of functional UGT1A1 enzyme. Previous reports on the clinical success of HT in CNI disease (4,10,11,19,27) prompted us to offer this therapy to the patient as a means to improve her quality of life (24 h home phototherapy required) and to prevent neurological complications. Fox et al. reported the case of a 10-year-old girl with CNI who was treated with HT (19), and this treatment lowered bilirubin levels and the hours of phototherapy needed and increased UGT1A1 activity.
Following six hepatocyte infusions to Patient 2, unconjugated bilirubin and skin jaundice considerably diminished, as described in previous cases (3,4,10,11,19,27), and the former daily phototherapy schedule was alleviated from 24 to 12 h. Dhawan et al. (11) reported two patients aged 18 months and 3 years in whom serum bilirubin was lowered over a 7-month follow-up period by 50% and 30%, respectively, after HT. Ambrosino et al. (4) also described decreased bilirubin levels, which were evident up to 3 months post-LT. In our child, we noted a gradual decrease and stabilization of unconjugated bilirubin that dropped by 50% after HT when compared to the initial values. This values have remained for the following (almost) 2 years with occasional cell infusions (Fig. 2). Of the eight patients with CNI that underwent LT worldwide, HT was indicated for seven of them as a bridge to LT, which was conducted a few months later (3,4,10,11,19,27). In our patient, we have achieved long-lasting stabilization of the serum bilirubin levels, which can be reasonably attributed to successfully transplanted and metabolically active hepatocytes. This achievement has not only significantly improved her quality of life but has also lowered the burden on the family thus helping all family members to lead a more normal life.
Several reports have been published about the indication of LT to treat GSD (1,6); however, considering the risk associated with LT and the prognosis of this illness, palliative medical treatment together with strict compliance of dietetic habits are the therapeutic measures recommended for the time being (9). In some cases, despite strict compliance of dietary measures, no reasonably metabolic control is achieved, which was the case of our patient. In these situations, HT could be considered as a therapeutic option. HT has been carried out in two adult patients with GSD-Ia and -Ib (von Gierke disease) and resulted in improved glucose control in both cases for at least 9 and 7 months, respectively (26,31). However, no further information is available about their evolution (26,31). HT was offered to P3 for the purpose of reducing hypoglycemia episodes. HT, as in the two previously reported cases, had a positive effect on this patient; the treatment was well tolerated with no adverse events over the following months, while the laboratory data indicated that lactic acid had dropped, fasting glucose levels were higher, and less hypoglycemia episodes occurred.
One of the mainstays of treatment of von Gierke's disease is the use of UCCS, which is given intermittently to provide stable blood glucose concentrations and to reduce swings of insulin secretion. Depending on the dose and frequency of UCCS, normoglycemia can be maintained for up to 6–9 h (54). Thus, it could be argued that the benefit observed in this patient was solely due to this dietary measure. Yet, this treatment failed to stabilize P3 glucose levels prior to HT, with frequent episodes of loss of consciousness, which made out-of-home activities, such as school attendance, very difficult. However after HT, there was a measurable correction of hypoglycemia, blood triglycerides lowered, as other authors reported (26,31), and there were no new episodes of loss of consciousness. The sustained reduction of lactic acid levels from 12 to 8 mmol/L suggests that at least some hepatocytes must be active in gluconeogenesis. This clinical improvement had as a consequence that the patient can now attend school regularly.
The clinical diagnosis of P4 was unsure, despite a strong clinical suspicion of tyrosinemia type I. The absence of succinylacetone in urine and the lack of response to NTBC and diet were intriguing and made uncertain the diagnosis and extensive genetic analysis, including full sequencing of the fumarylacetoacetate hydrolase gene, was requested. The genetic study, which took about one further month, confirmed the diagnosis of the disease and revealed the existence of mutations IVS6-1 G>T and IVS12+5G>A in this gene, both of which relate to a bad prognosis. This genetic analysis was further confirmed by a second, independent study.
When a metabolic disease is suspected in a patient, lack of an unequivocal diagnosis raises ethical concerns about the appropriateness and risk/benefit of LT. While genetic diagnosis was being performed for P4, we considered HT as a therapeutic option to slow down his metabolic deterioration until the definite diagnosis was made. Previous studies in fumarylacetoacetate hydrolase-deficient mice have demonstrated that a small number of transplanted differentiated adult hepatocytes could correct hepatic failure (34,35). Consequently, it was conceivable that P4 could benefit from HT.
Following HT, while maintaining the same diet and NTBC treatment as before, we recorded an increase in clotting factors and improved liver function, as evidenced by lower bilirubin levels. The progressive increase in clotting factors that occurred over 3 weeks (except in the course of a transient infection episode) cannot be simply attributed to the plasma transfusions administered 2 days before HT (Fig. 5), but to hepatocyte synthesis. Our patient is the first case reported to display a transient biochemical positive response after HT.
The acute form of this disease appears early in life with a poor response to treatment and death within the first year if diagnosis and treatment are not properly established early (15,28). The previous deterioration observed in P4 together with the identified genotype precluded a bad prognosis for his disease. Consequently and despite the initial satisfactory HT results, once the genetic study was confirmed, the patient was placed in the waiting list and underwent liver transplantation few days later. Our attitude toward this patient was guided by the fact that his clinical status was quite stable; had his condition worsened more rapidly or more severely, LT would have been decided even without a clear diagnosis and without further awaiting for a response to dietary and pharmacological treatment
Although imaging studies did not show portal flow impairment or ascites previous to HT, this patient showed unexpected transient alterations of portal flow following HT that could be managed satisfactorily and without clinic symptoms. By day 14 after HT, decreased portal flow was again observed, but coincidentally with a septic episode; ascites developed thereafter. Whether infusion of hepatocytes could have had a significant influence on ascites development by day 14, post-HT remains an open question. However, the fact that portal flow alterations normalized within hours after HT seems to minimize this possibility. Rather, the disease phenotype with rapid liver deterioration could justify the gradual alteration of portal hypertension, which is in agreement with the incipient liver cirrhosis that was later detected in the explanted liver. Thus, in similar cases in which liver cirrhosis /fibrosis can be suspected independently of histological or image evidence, indication for HT has to be carefully weighted.
In summary, our results reveal that patients with hepatic inborn metabolic diseases may benefit to a greater or lesser extent from hepatocytes cell therapy. In all cases, we obtained biochemical evidence that transplanted hepatocytes were metabolically active in the host for a limited period of time. The biochemical effects were particularly long-lasting in the CNI patient. To a certain extent, clinical improvement was observed in the treated patients, which is in agreement with previously published results (7,17,24,42,49,51). Although these results are encouraging, they do not suffice to ensure a resolutive outcome of HT in all the patients. HT stabilized the disease in P2 and P3, allowing a better quality of life. In P4, HT served as a bridge to LT, and likely it slowed down her clinical deterioration while she was on the liver transplant waiting list.
Footnotes
Acknowledgments
The authors are indebted to Fondo de Investigaciones Sanitarias and CIBERHED for their financial support. M.Á.C.A. was a recipient of a Bancaja-Fundación La Fe post-resident research contract. A.B-C. was supported by a Fundacion Lubasa grant. The authors declare no conflicts of interest.