
Abstract
The efficacy of islet transplantation is limited by poor graft vascularization. We herein demonstrated that microvesicles (MVs) released from endothelial progenitor cells (EPCs) enhanced human islet vascularization. After incorporation into islet endothelium and β-cells, EPC-derived MVs favored insulin secretion, survival, and revascularization of islets transplanted in SCID mice. MVs induced in vitro islet endothelial cell proliferation, migration, resistance to apoptosis, and organization in vessel-like structures. Moreover, MVs partially overcame the antiangiogenic effect of rapamycin and inhibited endothelial–leukocyte interaction via L-selectin and CD40. MVs were previously shown to contain defined patterns of mRNAs. Here we demonstrated that MVs carried the proangiogenic miR-126 and miR-296 microRNAs (miRNAs). MVs pretreated with RNase or derived from Dicer knocked-down EPCs showed a reduced angiogenic effect. In addition, MVs overcame the antiangiogenic effect of the specific antagomiRs of miR-126 and miR-296, suggesting a relevant contribution of miRNAs delivered by MVs to islet endothelium. Microarray analysis of MV-stimulated islet endothelium indicated the upregulation of mRNAs coding for factors involved in endothelial proliferation, differentiation, and angiogenesis. In addition, MVs induced the activation of the PI3K-Akt and eNOS signaling pathways in islet endothelium. These results suggest that MVs activate an angiogenic program in islet endothelium that may sustain revascularization and β-cell function.
Introduction
Islet transplantation is a suitable therapeutic option for type 1 diabetes (6,44). However, a great percentage of transplanted islets fail to engraft into the liver, and pancreata from multiple donors are still necessary to achieve insulin independence (6). A poor vascular engraftment is one of the main causes of islet loss. Therefore, the identification of factors able to enhance neoangiogenesis may increase the success of islet transplantation. It has been suggested that bone marrow-derived endothelial progenitor cells (EPCs) specifically localize within sites of endothelial injury and induce a regenerative program (49). Indeed, EPC infusion exerts protective effect in experimental models of hindlimb ischemia and myocardial infarction (2,43). Moreover, recipient EPCs are able to chimerize with donor vessels in transplanted organs, suggesting a putative role in graft revascularization (25,39). It has been previously shown that EPCs are recruited to the pancreas in response to islet injury and that EPC-mediated pancreas neovascularization may facilitate the recovery of injured β-cells, improving islet allograft function (29). In addition, the increase of EPCs in the peripheral circulation after granulocyte-macrophage colony-stimulating factor-induced mobilization resulted in an increase of vascular density and engraftment in a murine model of islet transplantation (17).
Several studies suggested that EPCs promote angiogenesis without a direct contribution in the formation of vessels, but rather by a paracrine mechanism (37). Exosomes/microvesicles (MVs) are thought to play an important role in cell-to-cell communication (12,30,40). We recently demonstrated that MVs released by EPCs are internalized into endothelial cells and activate an angiogenic program by horizontal transfer of mRNAs (18).
The aim of this study was to evaluate the effects of EPC-derived MVs on islet angiogenesis, insulin secretion, and islet endothelial–leukocyte interaction, all biological events crucial for transplanted islet engraftment and survival.
Materials and Methods
Human Islet and Islet-Derived Endothelial Cell Isolation
For the present study, we used islets discarded from the clinical transplant for insufficient islet mass. Authorization for islet purification from deceased donors was obtained from the Piedmont Region Healthcare and Italian National Transplant Centre (CNT). Ten different preparations of islets were prepared following the Ricordi method (41). Islet endothelial cell lines (IECs) were generated and characterized as previously described (4).
EPC Isolation and Characterization
EPCs were isolated from peripheral blood mononuclear cells (PBMCs) of healthy donors by density centrifugation, seeded on type I collagen-coated plates (48), and characterized on the basis of fluorescence-activated cell sorting (FACS) analysis showing the expression of stem and endothelial markers and of the uptake of 1,1-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-labeled acetylated low-density lipoprotein, as previously described (3,18).
IEC and EPC Lentiviral Infection
IECs and EPCs were transduced with a lentivector carrying a CMV-GFP (green fluorescent protein) expression cassette (13). Infected cells were termed IEC-GFP and EPC-GFP and characterized by FACS and immunofluorescence. The EPC-GFP cells were used only for experiments of microchimerism.
Islet–EPC Microchimerism
Microchimerism between islets [1,000 islet equivalent (IEQ)] and EPC-GFP (1 × 106 cells) was performed in microgravity by using the Rotary cell culture system (Synthecon, Houston, TX).
Isolation and Characterization of MVs From EPCs
MVs were obtained from supernatants of EPCs by ultracentrifugation (Optima L-90K, Beckman Coulter, Fullerton, CA) and characterized as previously described (18). Antigen expression on MVs was studied by FACS using antibodies directed to CD14, CD34, CD42b, L-selectin, P-selectin, CD154 (Dako, Copenhagen, Denmark), α4 integrin (Becton Dickinson, San Jose, CA), αVβ3 integrin, α6 integrin (BioLegend, San Diego, CA), and human leukocyte antigen (HLA) class I and II (Santa Cruz Biotech., Santa Cruz, CA). RNA extraction from MVs was performed using the mirVana isolation kit (Ambion, Austin, TX). RNA was analyzed by Agilent 2100 bioanalyzer (Agilent Tech. Inc., Santa Clara, CA). In situ hybridization was performed on MVs using a miRCURY LNA detection probe against microRNA-126 (miR-126) and miR-296 according to the manufacturer (Exiqon, Vedbaek, Denmark). The probe used was biotinylated at 5′ of its sequence and a scramble-miR probe (5′-biotin) was used as control (16). miR-126 and miR-296 expression levels were analyzed by qRT-PCR in a 48-well StepOne™ Real Time System (Ambion): 200 ng of RNA was reverse-transcribed and the cDNA was used to detect and quantify miR-126 and miR-296 by qRT-PCR using the miScript SYBR Green PCR Kit (Qiagen, Valencia, CA). In selected experiments, MVs were labeled with the red fluorescent dye PKH26 (Sigma Aldrich, St. Louis, MO) or treated with 1 U/ml RNase (Ambion) for 3 h at 37°C. After RNase treatment the reaction was stopped by addition of 10 U/ ml RNase inhibitor (Ambion) and MVs were washed by ultracentrifugation (18). The efficacy of RNase treatment was evaluated by MV-RNA analyses by Agilent 2100 bioanalyzer (Agilent). In addition, RNA extracted from RNase-treated and untreated MVs was labeled by oligo dT-driven retrotranscription and analyzed on 0.6% agarose gel showing the complete degradation of RNA by RNase (18).
Internalization of MVs in Human Islets and IECs
Islets (500 IEQ) cultured in microgravity and IECs seeded on six-well plates were incubated with 10 μg/ml labeled MVs. MV internalization was evaluated by confocal microscopy (Zeiss LSM 5 PASCAL, Jena, Germany) and FACS in the presence or absence of 1 μg/ml blocking antibodies directed to αVβ3 integrin (BioLegend), α4 integrin, α5 integrin (Chemicon Int., Temecula, CA), CD29, or L-selectin (Becton Dickinson). In selected experiments, IECs were engineered to knock-down CD40 by small interfering RNA (siRNA) (Santa Cruz Biotech.). MV internalization was also evaluated in the endothelial cell line H-END transfected with α(1,3/4)-fucosyltransferase (H-END fucosyl) or control vector (H-END cep) (7).
Assessment of Insulin Secretory Response, Islet Viability, and Caspase-3 Activity
Stimulation index (SI) was obtained from protein normalized (Bradford method) values of insulin secretion between low- and high-glucose islet challenge at different time points (5).
Control and MV-stimulated islets were challenged with Krebs-Ringer bicarbonate buffer (KRBB, pH 7.35) containing 0.5% bovine serum albumin (BSA; Sigma) under low-glucose concentration conditions (2.8 mM) and with high-glucose concentration (28 mM). Ten handpicked islets for each experimental group were incubated in a 12-well plate with 1 ml of KRBB plus low glucose (2.8 mM) for 60 min to allow the stabilization of insulin secretion. Islets were then incubated for 60 min in KRBB plus low glucose (2.8 mM) or in KRBB plus high glucose (28 mM) at 37°C and 5% CO2. Supernatants were collected under a microscope to avoid islet contamination and insulin quantification was performed by using an ELISA kit for human insulin (ALPCO, Windham, NH). Data are expressed as mean ± SD of five different experiments using five different preparations of human islets. Islet viability was assessed by dual staining with 0.46 μM fluorescein diacetate and 14.34 μM propidium iodide (Sigma Aldrich). The activity of caspase-3 was assessed by ELISA (Chemicon Int.).
PBMC Adhesion to IECs
PBMCs were isolated from healthy volunteers by density gradient and labeled with 10 μm Vybrant cell tracer (Invitrogen, Carlsbad, CA). Cells (50 × 106/ml) were added to IECs previously incubated with vehicle alone or 20 ng/ml tumor necrosis factor-α (TNF-α) and 20 ng/mL interferon-γ (IFN-γ) cytokines (Sigma Aldrich) in the presence or absence of 10 μg/ml MVs. PBMC adhesion to IECs was also performed in the presence of MVs pretreated with 1 μg/ml blocking antibody against L-selectin (Becton Dickinson) or with 1 μg/ml CD40-murine immunoglobulin (Ig) fusion protein (CD40 fp) (Alexis Bioch., San Diego, CA) (15).
Endothelial Outgrowth From Islets
Cell outgrowth from islets (500 IEQ) in the presence of vehicle alone or 10 μg/ml MVs was studied under a Nikon microscope system for living cell analysis. In selected experiments, rapamycin (10 ng/ml) or MVs pretreated with 1 U/ml RNase were used. A medium containing endothelial growth factors was used as positive control for cell outgrowth (14). Endothelial phenotype of outgrowing cells was confirmed by immunostaining for von Willebrand factor (vWF) (Dako), CD31, vascular endothelial growth factor receptor-2 (VEGFR2 or KDR), and CD105 (R&D Systems, Minneapolis, MN).
Xenografts in Severe Combined Immunodeficient (SCID) Mice
Islets (1,000 IEQ) or IEC-GFP (104 cells) were mixed to 500 μl Matrigel (Becton Dickinson) in the presence of vehicle alone or 10 μg/ml MVs pretreated or not with 1 U/ml RNase and implanted subcutaneously into the scruff region of the neck of SCID mice. After 7 days, mice were sacrificed and Matrigel plugs were retrieved for histology and immunohistochemistry. Six animals for each experimental group were examined. Animal studies were approved and conducted in accordance with the National Institute of Health guidelines.
In Vitro Assays on IECs
Angiogenesis
Formation of capillary-like structures was studied on IECs (5 × 104) seeded for 6 h on Matrigel and observed under an inverted microscope.
Migration
IEC migration was studied under an inverted microscope. The net migratory speed was calculated by the MicroImage software (Casti Imaging, Venice, Italy) based on the straight line distance between the starting and ending points divided by the time of observation (9). Migration of at least 30 cells for each experimental point was analyzed.
Proliferation
IECs (5 × 104) were cultured on 24-well plates and incubated with different stimuli and 10 μM bromodeoxyuridine (BrdU; Roche Diagnostics, Mannheim, Germany). The absorption values were determined at 405 nm wave length.
Apoptosis
IECs were subjected to terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (Chemicon Int.). Samples were analyzed under a fluorescence microscope and green-stained apoptotic cells were counted in 10 nonconsecutive microscopic fields. In all assays, IECs were incubated with 10 μg/ml MVs in the presence or absence of 10 ng/ml rapamycin, 0.1 mM wortmannin, 10 μM l-nitro-arginine methyl ester (L-NAME), or 10 μM d-nitro-arginine methyl ester (D-NAME) (all from Sigma Aldrich). In selected experiments MVs were pretreated with 1 U/ml RNase.
Gene Array Analysis
Human GEarray kit for the study of angiogenesis markers (SuperArray Inc., Bethesda, MD) was used to characterize the gene expression profile of IECs incubated with vehicle alone versus 10 μg/ml MVs for 48 h. Microarray data are deposited on European Bioinformatic Institute (EBI) website (http://www.ebi.ac.uk/microarray-as/ae/) as E-MEXP-2511 (experiments archive code).
Western Blot Analysis
IEC protein lysates (30 μg) were subjected to 4–15% gradient sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto nitrocellulose membranes. Primary antibodies directed to Akt (Upstate, Charlottesville, VA), phosphorylated (P)-Akt and P-eNOS (endothelial nitric oxide synthase; Cell Signalling, Beverly, MA), eNOS, and actin (Santa Cruz Biotech.) were used.
Knock-Down of Dicer in EPCs
Knock-down of Dicer in EPCs was performed by a specific siRNA according to the manufacturer (Santa Cruz Biotech.). An irrelevant siRNA was used as control. Western blot for Dicer expression was performed by using an anti-Dicer polyclonal antibody (Abcam, Cambridge, UK). MVs were collected by supernatants of engineered cells at day 4 after transfection and used for proliferation and angiogenesis assays on IECs.
Transfection of IECs with miR-126 and miR-296 AntagomiRs
Oligonucleotide inhibitors (100 pmol) (antagomiRs, anti-miR-126, and anti-miR-296) (Ambion) were transfected into IECs seeded on six-well plates by using 4 μl lipofectamines 2000 (Invitrogen). IECs were also transfected with a predesigned 5-carboxyfluorescein (FAM)-conjugated anti-miR without biological activity as control. AntagomiR-transfected IECs were incubated with 10 μg/ml MVs in proliferation and angiogenesis assays.
Statistical Analysis
All data of different experimental procedures are expressed as average ± SD. Statistical analysis was performed by Student's t-test or ANOVA with Newmann-Keuls or Dunnet's multicomparison test where appropriated. Kolmogorov-Smirnov statistical nonparametric test was performed to evaluate the significance of FACS analysis.
Results
Human Islets Formed Chimeric Micro-organoids with EPCs
Human islets were cocultured in microgravity with GFP-expressing EPCs obtained from peripheral blood of six different healthy volunteers. EPC-GFP adhered to islet surface and grew, forming chimeric micro-organoids containing dithizone-stained islets (Fig. 1A, B). EPC–islet chimeras showed a significant increase of insulin secretion compared to islets alone (SI 2.83 ± 0.67 EPC–islet chimeras vs. 1.87 ± 0.32 islets alone at day 2). Similar results were obtained incubating islets with supernatants of EPCs (SI 3.02 ± 0.81 islet–EPC supernatants vs. 1.94 ± 0.51 islets alone at day 2), suggesting a paracrine effect. The use of lentivirus-infected EPC-GFP were limited only to the experiments of microchimerism.

Formation of human islet–EPC (endothelial progenitor cell) chimeric micro-organoids and characterization of EPC-derived microvesicles (MVs). (A, B) Representative micrographs showing the formation of chimeras between green fluorescent protein (GFP)-expressing EPCs and human islets in microgravity culture (A). Islets presented a maintained dithizone staining (B) and are surrounded by EPCs after 2 days of culture [original magnification (A, B): ×100; scale bar: 100 μm]. (C–E) Transmission (C, D) and scanning (E) electron microscope analysis of MVs showing a spheroid shape [original magnification: (C) ×10,000, (D) ×25,000, (E) ×20,000; scale bar: (C) 500 nm, (D, E) 100 nm]. Digital acquisition and analysis were performed using the JEOL Semafore system. (F) Fluorescent-activated cell sorting (FACS) analysis of surface antigens expressed on MVs (filled curves) showing the presence of α4 and β1 integrins, CD34, CD154, and L-selectin and the absence of human leukocyte antigen (HLA) class I antigens and of platelet (CD42b, P-selectin) or monocyte (CD14) markers. Control-matched isotype was used as experimental control (open curves). Kolmogorov-Smirnov statistical analysis was performed. (G) Bioanalyzer profile of RNA subtypes present in EPCs and EPC MVs showing an enrichment for small RNAs in MVs. (H–J) In situ hybridization of MVs for microRNA-126 (miR-126) (H), miR-296 (I), or control scramble miR (J) [original magnification: (H–J) ×25,000; scale bar: 50 nm]. (K) Representative qRT-PCR analysis for miR-126 (1 and 3) and miR-296 (2 and 4) in MVs stimulated with vehicle alone (1 and 2) or pretreated with 1 U/ml RNase (3 and 4).
Characterization of EPC-Derived MVs
We investigated whether MVs obtained from the supernatants of EPCs may account for this paracrine effect. Transmission (Fig. 1C, D) and scanning (Fig. 1E) electron microscopy showed the spheroid morphology of MVs. Whereas a small percentage of MVs (8–10%) showed a size around 1 μm, the majority of them ranged between 60 and 130 nm. By FACS analysis, MVs were detectable under the 1-μm beads and expressed the hematopoietic stem cell marker CD34 and molecules essential for leukocyte adhesion (3) such as α4 and β1 integrin, CD154 (CD40-L), and L-selectin (Fig. 1F). MVs did not express HLA class I and class II and markers of platelets (P-selectin, CD42b) and monocytes (CD14) (Fig. 1F). MVs carried different RNAs enriched in small RNAs including miRNAs as seen by bioanalyzer (Fig. 1G). By in situ hybridization (Fig. 1H–J) and qRT-PCR (Fig. 1K) the proangiogenic miR-126 and miR-296 were detected within MVs. As seen in Figure 1K, RNase pretreatment of MVs abated the expression of miR-126 and miR-296.
MV Internalization in β-Cells Sustained Insulin Secretion and Viability
Confocal microscopy analysis showed the internalization of PKH26-labeled MVs into β-cells as detected by costaining with insulin and glucose transporter-2 (GLUT-2) (Fig. 2A). However, internalization was not β-cell specific as MVs were also detectable in glucagon-positive cells (Fig. 2A). Islet function, evaluated as insulin secretion by high-glucose challenge 2 and 7 days after MV stimulation, was significantly higher in respect to vehicle alone (Fig. 2B). In addition, dual staining with fluorescein diacetate and propidium iodide evidenced an enhanced islet viability in the presence of MVs (92.45 ± 7.56% islets + MVs vs. 80.35 ± 6.45% islets alone at day 2). The inhibition of islet apoptosis induced by MVs was confirmed by the significant decrease of caspase-3 activity (Fig. 2C).

Internalization of MVs in islet endothelium and β-cells and effect on insulin secretion and islet apoptosis. (A) Confocal microscopy analysis of red-labeled MVs (MV PKH26) in β-cells and islet endothelium after staining (green) for insulin, glucose transporter-2 (GLUT-2), glucagon, CD31, or vascular endothelial growth factor receptor 2 (VEGFR2). Scale bar: 50 μm. (B) Significant increase of insulin secretion (ELISA) induced by MVs in islets after 2 and 7 days of culture (*p < 0.05 MVs—white bars vs. vehicle alone—black bars). (C) Significant decrease of apoptosis (caspase-3 ELISA) induced by MVs in islets after 2 and 7 days of culture (*p < 0.05 MVs—white bars vs. vehicle alone—black bars). (D, E) Confocal microscopy (D) and FACS (E) analysis of red-labeled MV internalization in islet endothelial cells (IECs). In (D), red-labeled MVs are visible in IEC cytoplasm (lower panel) in respect to unstimulated control (upper panel). Nuclei were counterstained by 0.5 μg/ml Hoechst (original magnification: ×400; scale bar: 10 μm). In (E) left upper panel, representative FACS analysis of PKH26-labeled MV internalization in IECs (black-filled curve) in respect to cells incubated with vehicle alone as internal control (white-filled curve). Preincubation with 1 μg/ml blocking antibody against α4 integrin (left lower panel), β1 integrin (right upper panel), or L-selectin (right lower panel) inhibited MV internalization in IECs. Gray-filled curves indicate the MVs internalized in the absence of blocking antibodies, whereas black-filled curves indicate the MVs internalized after incubation with blocking antibodies. (F) FACS analysis of PKH26-labeled MV internalization in IECs engineered to knock-down CD40 (lower panel) or with a control siRNA (upper panel). (G) FACS analysis of PKH26-labeled MV internalization in the endothelial cell line H-END transfected to express surface L-selectin ligands (H-END fucosyl, upper panel) or with a control empty plasmid (H-END cep, lower panel). In (F) and (G), black-filled curves indicate MV internalized in comparison to cells stimulated with vehicle alone (white-filled curves). Three experiments were performed with similar results and Kolmogorov-Smirnov statistical analysis was performed.
MVs Were Internalized in Islet Endothelium via α4/β1 Integrins, L-Selectin, and CD40
Confocal microscopy analysis revealed that MVs were internalized into islet endothelium as assessed by costaining with the endothelial markers CD31 and VEGFR2 (KDR) (Fig. 2A). These results were confirmed by incorporation of MVs in different lines of isolated IECs as shown by confocal microscopy (Fig. 2D) and FACS (Fig. 2E). The selective blockade of α4 integrin, β1 integrin, and L-selectin induced a significant decrease of MV internalization into IECs (Fig. 2E). Moreover, MV internalization was decreased in IECs engineered to knock-down CD40 by specific siRNA (Fig. 2F) and in an endothelial cell line (H-END) not expressing fucosylated residues that bind to L-selectin (Fig. 2G).
MVs Inhibited PBMC Adhesion to IECs
The addition of 10 μg/ml MVs significantly inhibited spontaneous and cytokine-induced PBMC adhesion to IECs (Fig. 3A). A fusion protein able to inhibit CD40–CD154 interaction (CD40fp) or a blocking monoclonal antibody directed to L-selectin significantly reduced PBMC adhesion (Fig. 3B). Moreover, MVs significantly reduced PBMC adhesion to the endothelial cell line (H-END) engineered to overexpress L-selectin ligands (Fig. 3B). These results suggest a crucial role for CD40 and L-selectin expressed on MVs in the inhibition of PBMC adhesion to IECs.

Effects of MVs on endothelial–leukocyte interaction and on endothelial cell outgrowth from islets. (A) Effect of MVs on spontaneous and cytokine-induced adhesion of peripheral blood mononuclear cells (PBMCs) to IECs. Labeled PBMCs were added to IECs incubated with vehicle alone, 10 μg/ml MVs, 20 ng/ml tumor necrosis factor-α (TNF-α) plus 20 ng/ml interferon-γ (IFN-γ) (cytokines) in the presence or absence of 10 μg/ml MVs. MVs significantly reduced spontaneous (*p < 0.05 MV vs. vehicle alone) and cytokine-induced (*p < 0.05 cytokines vs. vehicle and #p < 0.05 cytokines + MV vs. cytokines) PBMC adhesion. (B) Role of CD40 and L-selectin on PBMC–IEC interaction: labeled PBMCs were added to IECs in the presence of vehicle alone, 10 μg/ml MVs, 10 μg/ml MVs + 1 μg/ml CD40-murine Ig fusion protein (MV + CD40 fp), or 10 μg/ml MVs + 1 μg/ml blocking antibody directed to L-selectin (MV + anti-L-selectin). CD40 fp and L-selectin blocking antibody significantly inhibited MV-induced decrease of PBMC adhesion to IECs (*p < 0.05 MV + CD40 fp or MV + anti-L-selectin vs. MV). MVs also induced a significant decrease of PBMC adhesion to the endothelial cell line H-END transfected with an empty control vector (H-END cep) (#p < 0.05 H-END cep + MV vs. H-END cep). MV-induced inhibition of PBMC adhesion was more evident in H-END transfected with α(1,3/4)-fucosyltransferase (H-END fucosyl) to induce the surface upregulation of oligosaccharides residues that bind to L-selectin (†p < 0.05 H-END fucosyl + MV vs. H-END fucosyl). In (A) and (B), all samples were fixed and analyzed under a UV light microscope. Fluorescent cells in 10 fields/well were counted at ×200 magnification and data are representative of average number of adherent cells/field ± SD. Three independent experiments were performed with similar results. (C–K) Analysis of cell outgrowth from islets. (C–E) Representative micrographs (original magnification ×100; scale bar: 100 μm) of islets incubated with vehicle alone (C) or 10 μg/ml MVs for 24 h (D) or 72 h (E). (F) Representative micrograph (original magnification: ×200; scale bar: 100 μm) of islets cultured with 10 μg/ml MVs for 72 h on Matrigel-coated surfaces. (G–J) Representative micrographs of the endothelial phenotype of cells outgrowing from islets by immunostaining for von Willebrand factor (vWF) (G), VEGFR2 (H), CD31 (I), and CD105 (J). In all images, nuclei were counterstained by 1 μg/ml propidium iodide (original magnification: ×400; scale bar: 10 μm). (K) Count of area of cells outgrowing from islets by image software analysis of 10 nonconsecutive representative micrographs (magnification: ×100). MVs induced a significant increase of cell outgrowth (*p < 0.05 MV vs. vehicle). Coincubation of MVs with rapamycin (10 ng/ml) or pretreatment of MVs with 1 U/ml RNase significantly reduced cell outgrowth from islets (#p < 0.05 MV + rapamycin or MV + RNase vs. MV). The inhibition of cell outgrowth induced by rapamycin was significantly lower than that observed after RNase pretreatment of MVs (†p < 0.05 MV + RNase vs. MV + rapamycin). Rapamycin completely inhibited cell outgrowth from islets induced by a medium containing endothelial growth factors (EndoGF) used as experimental control. Similar results were obtained using islets isolated from five different deceased donors.
MVs Induced Endothelial Outgrowth From Islets
In comparison to vehicle alone (Fig. 3C), 10 μg/ml MVs induced cell outgrowth from islets detectable after 24 h (Fig. 3D) and enhanced after 96 h (Fig. 3E). Cells outgrowing from islets formed capillary-like structures on Matrigel-coated plates (Fig. 3F) and showed a typical endothelial phenotype expressing vWF (Fig. 3G), VEGFR2 (Fig. 3H), CD31 (Fig. 3I), and CD105 (Fig. 3J).
Effect of Rapamycin and RNase Pretreatment on MV-Induced Endothelial Outgrowth From Islets
Rapamycin (10 ng/ml) completely abrogated the islet endothelial outgrowth stimulated by growth factor-enriched medium (Fig. 3K). By contrast, the same dose of rapamycin did not completely abolish MV-induced endothelial outgrowth (Fig. 3K). In addition, pretreatment of MVs with 1 U/ml RNase markedly inhibited endothelial cell outgrowth, suggesting a horizontal RNA transfer (Fig. 3K).
MVs Induced an Early Vascularization of Human Islets Xenotransplanted in SCID Mice
The effect of MVs on islet neoangiogenesis was evaluated in vivo after subcutaneous xenotransplantation of islets within Matrigel in SCID mice (14). In the presence of MVs, transplanted islets showed an early (7 days) increase of vascular density as detected by hematoxylin/ eosin and CD31 immunostaining (Fig. 4A). The evaluation of total area of neoformed vessels within islets confirmed a significant increase of angiogenesis after MV stimulation (Fig. 4B). Pretreatment of MVs with 1 U/ml RNase significantly reduced islet revascularization (Fig. 4A, B). Insulin staining was maintained in all experimental conditions (Fig. 4A).

MVs induced an early vascularization of islets after xenotransplantation in severe combined immunodeficient (SCID) mice. (A) Hematoxylin/eosin staining and confocal microscopy immunostaining for the endothelial marker CD31 (red) and insulin (green) of human islets xenotransplanted subcutaneously into the scruff region of the neck of SCID mice after inclusion into Matrigel. MVs induced an early vascularization (7 days after transplantation) not detectable in islets incubated with MVs pretreated with 1 U/ml RNase or with vehicle alone. Insulin staining was maintained in all experimental conditions (original magnification ×250; scale bar: 100 μm). (B) The enhancement of MV-induced intraislet microvascular density was confirmed by the count of total area (μm2/section) of neoformed vessels in 10 nonconsecutive sections (*p < 0.05 MV vs. vehicle alone). RNase pretreatment reduced the proangiogenic effect of MVs (#p < 0.05 MV + RNase vs. MV). Six animals for each experimental group were used.
MVs Enhanced In Vivo and In Vitro IEC Angiogenesis
In the presence of MVs, IEC-GFP xenografts in SCID mice (14) showed a marked enhancement of proliferation and formation of neovessels as detected by hematoxylin/eosin staining (Fig. 5A) and fluorescence analysis (Fig. 5B). Moreover, MVs induced a significant increase of total area of vessels formed within Matrigel (Fig. 5C). The in vivo IEC angiogenesis was significantly inhibited by pretreatment of MVs with 1 U/ml RNase (Fig. 5A, C).

MVs enhanced in vivo IEC angiogenesis. (A, B) Hematoxylin/eosin staining (A) and fluorescence microscopy analysis (B) of IECs infected by a lentiviral vector carrying GFP and xenotransplanted subcutaneously into the scruff region of the neck of SCID mice after inclusion into Matrigel. MVs enhanced the formation of neovessels by IECs within Matrigel plugs (7 days after transplantation). This proangiogenic effect was reduced when MVs were pretreated with 1 U/ml RNase. (C) The enhancement of MV-induced IEC angiogenesis was confirmed by the count of total area (μm2/section) of neoformed vessels in 10 nonconsecutive sections (*p < 0.05 MV vs. vehicle alone). RNase pretreatment significantly reduced the proangiogenic effect of MVs (#p < 0.05 MV + RNase vs. MV). Six animals for each experimental group were used. Scale bar: 50 μm.
In vitro, IEC-GFP spontaneously formed capillary-like structures on Matrigel (Fig. 6A). The addition of MVs accelerated and enhanced this process (Fig. 6B). RNase pretreatment significantly reduced the number of MV-induced capillary-like structures (Fig. 6C, D). Moreover, rapamycin (10 ng/ml) did not completely abrogate the angiogenic effect of MVs (Fig. 6D): the phosphoinositide 3-kinase (PI3K) inhibitor wortmannin (0.1 mM) and the NOS inhibitor L-NAME (10 μM) but not the inactive enantiomer D-NAME (10 μM) significantly reduced the number of capillary-like structures induced by MVs (Fig. 6D).

MVs enhanced in vitro IEC angiogenesis, migration, and resistance to apoptosis. (A–D) Representative micrographs (A, C) and count of capillary-like structures (D) formed by IECs seeded on Matrigel-coated plates (angiogenesis assay). In comparison to vehicle alone (A, D), MVs (B, D) induced a significant increase of capillary-like structures (*p < 0.05 MV vs. vehicle alone). Pretreatment with 1 U/ml RNase (C, D) significantly reduced MV-induced angiogenesis (#p < 0.05 MV + RNase vs. MV). Scale bar: 10 μm. (D) The mTOR inhibitor rapamycin (10 ng/ml), the phosphate inositol 3 kinase (PI3K)-inhibitor wortmannin (0.1 mM), and the nitric oxide synthase (NOS) inhibitor L-NAME (10 μM) significantly reduced the angiogenic effect of MVs (#p < 0.05 MV + rapamycin, MV + wortmannin, or MV + L-NAME vs. MV). Incubation of IECs with the inactive enantiomer D-NAME (10 μM) did not alter MV angiogenic effect. The mTOR, PI3K, and NOS pharmacologic inhibition did not reach the reduction of the angiogenic effect observed by pretreatment of MVs with RNase but the difference was not statistically significant. (E, F) Effect of MVs on migration (E) and inhibition of IEC apoptosis induced by serum deprivation (F). In comparison to vehicle alone, MVs induced a significant increase of migration (E) and inhibition of apoptosis (F) (*p < 0.05 MV vs. vehicle alone). Pretreatment with 1 U/ml RNase significantly reduced MV-induced migration (E) and inhibition of apoptosis (F) (#p < 0.05 MV + RNase vs. MV). The mTOR inhibitor rapamycin (10 ng/ml), the PI3K inhibitor wortmannin (0.1 mM), and the NOS inhibitor L-NAME (10 μM) significantly reduced the migratory (E) and antiapoptotic (F) effects of MVs (#p < 0.05 MV + rapamycin, MV + wortmannin, or MV + L-NAME vs. MV). Incubation of IECs with the inactive enantiomer D-NAME (10 μM) did not alter MV-induced migration and inhibition of apoptosis. The mTOR, PI3K, and NOS pharmacologic inhibition did not reach the reduction of migration and resistance to apoptosis observed by pretreatment of MVs with RNase († p < 0.05 MV + RNase vs. MV + rapamycin, MV + wortmannin, or MV + L-NAME). For all assays, three independent experiments were performed with similar results.
MVs Exerted Migratory, Proliferative, and Antiapoptotic Effects on IECs
The baseline migration rate of IECs studied by time-lapse recording microscopy remained stable for the whole period of observation never exceeding 5–6 μm/h (Fig. 6E). MVs significantly increased cell motility (Fig. 6E). In addition, MVs significantly increased IEC proliferation (not shown) and inhibited apoptosis (Fig. 6F). RNase pretreatment (1 U/ml) abrogated such effects (Fig. 6D, F). The incubation with rapamycin, wortmannin, and L-NAME but not D-NAME significantly decreased MV-induced IEC angiogenesis (Fig. 6D), migration (Fig. 6E), proliferation (not shown), and apoptosis resistance (Fig. 6F).
Pathways Involved in MV-Induced IEC Angiogenesis
MVs enhanced the IEC gene expression of endothelial differentiation sphingolipid G-protein-coupled receptor (EDG-1), tyrosine kinase receptor ephrin B4, fibroblast growth factor-receptor (FGF-R), VEGF-R2, transforming growth factor-β-receptor (TGF-β-R), α5 and β3 proangiogenic integrins, fibronectin-1, CD31, and tie-2 specific endothelial markers and eNOS. In addition, MV-stimulated IECs showed the downregulation of the antiangiogenic factors thrombospondin-1 (TSP-1) and 2 (TSP-2) (Fig. 7A). Western blot analysis showed the phosphorylation of Akt and the enhanced expression and phosphorylation of eNOS in IECs incubated with MVs (Fig. 7B).

Gene array analysis of MV-stimulated IECs and activation of the P-Akt/Akt and P-eNOS/eNOS pathways. (A) Gene array profiling of IECs incubated with MVs versus vehicle alone (angiogenesis-related genes). The graph shows the fold variation of angiogenesis-related genes between IECs stimulated with MVs versus vehicle alone. Samples were normalized for the signals found in housekeeping genes [actin, glyceraldehyde 3 phosphate dehydrogenase (GAPDH)]. Three independent experiments were performed with similar results. Gene table: EDG-1, endothelial differentiation sphingolipid receptor; EPHB4, ephrin B4; ETS-1, transcriptional factor c-ets; FGFR1, fibroblast growth factor receptor 1; KDR (FLK-1), vascular endothelial growth factor receptor 2; FN1, fibronectin 1; ITGA5, integrin, α5; ITGAV, integrin, αV; ITGB3, integrin, β3; AMOT, angiomotin; MADH1, Smad1 (transforming growth factor-β signaling protein 1); MDK, midkine (neurite growth-promoting factor 2); NOS3, nitric oxide synthase 3 (endothelial cell); PECAM, platelet/endothelial cell adhesion molecule (CD31 antigen); SPP1, osteopontin; TEK, Tie-2; TGFBR2, transforming growth factor, β receptor II (70–80 kDa); THBS1, thrombospondin 1; THBS2, thrombospondin 2. (B) Representative Western blot analysis of P-Akt, Akt, P-eNOS, and eNOS expression in IECs incubated with vehicle alone (lanes 1 and 3) or 10 μg/ml MVs (lanes 2 and 4). The graph shows the P-Akt/Akt ratio in the different experimental conditions. Three independent experiments were performed with similar results.
Role of miRNAs in MV-Induced IEC Angiogenesis
To study the role of miRNAs in MV-induced IEC angiogenesis, we engineered EPCs to knock-down Dicer, the intracellular enzyme essential for miRNA synthesis (Fig. 8). We found that MVs derived from Dicer siRNA-EPCs but not control siRNA-EPCs showed a significant decrease of proliferative (Fig. 8A) and angiogenic (Fig. 8B) effects on IECs. For these experiments, we used MVs derived from EPCs with the maximal knock-down of Dicer 4 days after transfection (Fig. 8C). qRT-PCR analysis showed reduced levels of miR-126 and miR-296 in MVs derived from siRNA Dicer but not from control siRNA EPCs (Fig. 8D). IECs were transfected with selective antagomiRs (antagomiR-126 or antagomiR-296) or a FAM-conjugated control antagomiR (Fig. 8E). A-miR-126 and A-miR-296 significantly reduced IEC proliferation (Fig. 8F) and angiogenesis (Fig. 8G). No effect was detected by using the FAM-conjugated control antagomiR (not shown). The addition of MVs overcame the inhibitory effects of both antagomiRs (Fig. 8E, F).
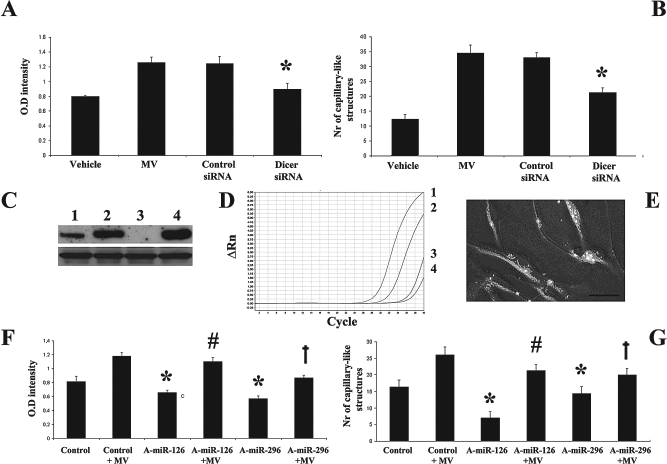
Role of miRNAs shuttled by MVs on IEC proliferation and angiogenesis. (A, B) Proliferation (A) and angiogenesis (B) assays of IECs incubated with vehicle alone, MVs derived from wild-type EPCs (MV), MVs derived from EPCs transduced with a control siRNA (control siRNA), or MVs derived from EPCs engineered to knock-down Dicer (Dicer siRNA). In comparison to MVs or control siRNA MVs, Dicer siRNA MVs showed a significant decrease of proliferative (A) and angiogenic (B) effect on IECs (*p < 0.05 Dicer siRNA vs. control siRNA or MV). (C) Representative Western blot analysis of Dicer (upper lane) and β-actin (lower lane) expression in EPCs subjected to Dicer siRNA (lanes 1 and 3) or control siRNA (lanes 2 and 4) 1 day (lanes 1 and 2) or 4 days (lanes 3 and 4) after transfection. (D) Representative qRT-PCR analysis for miR-126 (1 and 3) and miR-296 (2 and 4) in MVs derived from EPCs transfected with control siRNA (1 and 2) or Dicer siRNA (3 and 4). (E) Representative fluorescence micrograph showing the FAM-conjugated control antagomiR transfected into IECs (original magnification: ×400; scale bar: 10 μm). (F, G) Proliferation (F) and angiogenesis (G) assays of IECs transfected with specific antagomiRs directed to miR-126 (A-miR-126) or miR-296 (A-miR-296). A-miR-126 and A-miR-296 induced a significant reduction of IEC proliferation (F) and angiogenesis (G) (*p < 0.05 A-miR-126 or A-miR-296 vs. control). The addition of MVs to A-miR-stimulated IECs induced a significant increase of proliferation (F) and angiogenesis (G) overcoming the effect of the respective antagomiR (#p < 0.05 A-miR-126 + MV vs. A-miR-126 and † p < 0.005 A-miR-296 + MV vs. A-miR-296).
Discussion
In this study, we demonstrated that EPC-derived MVs enhanced transplanted islet neoangiogenesis.
Several studies addressed the relevance of angiogenesis in the early events occurring after intraportal infusion of islets. Purified islets maintain viable native endothelial cells able to trigger the revascularization by forming chimeric structures with the recipient vessels (10). Endogenous and exogenous factors may influence islet endothelium viability (4,34). Several proangiogenic growth factors (19,45) and composite islet–endothelial cell grafts were shown to improve islet vascularization and to inhibit the inflammatory response (23,24).
We evaluated whether EPCs were capable to form chimeric micro-organoids with islets. EPCs have been shown to be protective on ischemia-reperfusion injury and cardiovascular diseases (21,28). The origin of EPCs is still matter of debate. Some authors showed that the proangiogenic potential of these cells derived from the circulation may be due to the contamination with monocytes and platelet-derived products (38). To avoid contamination, we used MVs isolated from EPCs after three passages in culture. The cells used and the derived MVs expressed markers of stem cells (CD34, CD133) and of endothelium (VEGFR2, CD31) but not of monocytes (CD14) and platelets (P-selectin, CD41, CD42b).
It has been suggested that EPCs do not act via a direct trans-differentiation into mature endothelial cells, but rather by secreting angiogenic growth factors or other active molecules (46). The exogenous infusion of EPCs or their mobilization from the bone marrow favored the recovery of damaged β-cells in murine models of islet transplantation (17,29). Here we found that EPCs in microgravity formed with islets micro-organoids with enhanced viability, insulin secretion, and angiogenic properties. Since EPC supernatants mimicked the same effects on islets, it is conceivable that paracrine factors secreted from EPCs play a role. Recent studies suggested a paracrine action of MVs that may act as mediators of cell-to-cell communication (12). We previously demonstrated that MVs released from EPCs trigger angiogenesis by a horizontal transfer of mRNA (18). MVs are released by several cell types including platelets and cells of the hematopoietic lineage such as B and T lymphocytes, monocytes, and dendritic cells and display pleiotropic effects (1,8,20,32,35,36,42).
Herein we demonstrated that EPC-derived MVs are internalized in β-cells and islet endothelium sustaining insulin secretion and angiogenesis. MV uptake was mediated by specific cell membrane receptors such as α4 and β1 integrins, L-selectin, and CD154 (CD40-Ligand). We previously demonstrated that EPCs express L-selectin and that this molecule is essential for their homing at the sites of vascular injury (3). Here we showed that EPC-derived MVs internalized into target endothelial cells via L-selectin by binding to fucosylated residues or other oligosaccharidic ligands known to be upregulated after ischemia-reperfusion injury (11). Moreover, MVs expressed CD154 able to bind the costimulatory molecule CD40 which was expressed by IECs (14). These receptors not only mediated the internalization of MVs but, by competing with the molecules expressed on leukocytes, may interfere with leukocyte adhesion to endothelium. This could limit the triggering of inflammation and/or allo- and autoimmmune-mediated injury in transplanted islets.
We also found that MVs enhanced in vitro proliferation, resistance to apoptosis, migration of IECs, and endothelial outgrowth from islets. Gene array analysis of IECs stimulated with MVs showed an increased expression of angiogenesis-related molecules. In particular, MVs triggered the activation of PI3K/Akt and eNOS signaling pathways. MVs released by EPCs were previously shown to shuttle specific mRNAs associated with PI3K/Akt and eNOS pathways (18). MVs also induced in IECs the upregulation of ephrin B4 and EDG-1. Ephrins and their relative tyrosine kinase receptors have been identified in islet endothelium and were deeply involved in cell motility and adhesion (26). EDG-1 is the receptor for sphingosine 1-phosphate (S1P), which is known to activate the endothelial isoform of eNOS, thus inducing angiogenesis (22). Moreover, MV-stimulated IECs showed increased levels of CD31 (platelet/endothelial cell adhesion molecule 1; PECAM-1), a molecule known to inhibit endothelial apoptosis, and a reduced expression of TSP-1. TSP-1 is an inhibitor of angiogenesis, which also promotes apoptosis in activated endothelial cells (27). We have previously shown that TSP-1 is upregulated in IECs in response to rapamycin (14). Moreover, TSP-1 knockout mice presented islet hyperplasia characterized by an increased blood vessel density (33).
The significant reduction of MV activities after treatment with RNase suggests a putative horizontal transfer of RNAs. We previously characterized the mRNAs shuttled by EPC-derived MVs (18). In the present study, we found that MVs were enriched in the angiogenic miR-126 and miR-296 miRNAs (47). miRNAs are small noncoding RNAs able to modulate protein transduction (31). The role of miRNAs shuttled by MVs in islet angiogenesis was inferred by experiments with MVs derived from EPCs previously subjected to the knock-down of Dicer. Dicer is an intracellular enzyme essential for miRNA production (30). In Dicer-silenced MVs, the expression of the angiogenic miR-126 and miR-296 was decreased and the proliferation and angiogenesis of IECs was significantly reduced. On the other hand, MV-containing miR-126 and miR-296 were able to overcome the inhibitory effect of their specific antagomiRs. These results suggest that miRNAs shuttled by MVs contribute to their angiogenic effect.
In conclusion, MVs released from EPCs induced a protective effect on human islets, triggering neoangiogenesis. MVs represent a new concept of intercellular cross-talk through the transfer of biological products such as proteins, mRNAs, and miRNAs. Indeed, MVs may mimic the effect of the cells from which they are released. Further studies are needed to evaluate whether EPC-derived MVs may find a therapeutic application in preventing ischemia-reperfusion injury in solid organ and cell transplantation.
Footnotes
Acknowledgments
This work was supported by Italian Government Miur PRIN project, Regione Piemonte, Piattaforme Biotecnologiche PiSTEM project, Converging Technologies Nano IGT, and Ricerca Finalizzata and Local University Grants (‘ex60%). V.C., M.C.D., and G.C. are named inventors in a related patent (publication number: WO/2009/050742).