
Abstract
We sought to assess the potential of human cord blood-derived mesenchymal stem cells (CB-MSCs) to derive insulin-producing, glucose-responsive cells. We show here that differentiation protocols based on stepwise culture conditions initially described for human embryonic stem cells (hESCs) lead to differentiation of cord blood-derived precursors towards a pancreatic endocrine phenotype, as assessed by marker expression and in vitro glucose-regulated insulin secretion. Transplantation of these cells in immune-deficient animals shows human C-peptide production in response to a glucose challenge. These data suggest that human cord blood may be a promising source for regenerative medicine approaches for the treatment of diabetes mellitus.
Keywords
Introduction
Current treatments for type 1 diabetes mellitus are based on exogenous administration of insulin via multiple injections and/or delivery through microinfusors. While effective in maintaining reasonable blood glucose control, these treatments often fail to prevent hyper- and hypoglycemia, potentially resulting in acute events (hypoglycemic episodes) and/or chronic complications. Islet transplantation can restore physiological blood glucose control and eliminate severe hypoglycemia, improving quality of life (47).
As one of the outstanding issues that characterize islet transplantation is the scarcity of donors, the use of stem cell-derived insulin-producing cells represents a desirable goal. The hope of obtaining an unlimited source of cells for regenerative medicine was catalyzed by the isolation of human embryonic stem cells (hESCs) (55) and adult pluripotent mesenchymal stem cells (MSCs) (25). Both cell types have therefore received a great deal of attention as possible sources of insulin-producing cells (1,15,21,27,39,40,43,51,57,59). Adult stem cells are obtained from multiple tissues and are relatively simple to expand under defined conditions. In this respect, the stem cell-rich human umbilical cord blood (CB) represents a promising source for the treatment of insulinopenic diabetes.
We therefore explored the use of human CB-MSCs as a source of insulin-producing cells through in vitro differentiation with defined stepwise culture protocols. Selected markers that are seen in pluripotent stem cells [octamer-binding transcription factor 4 (OCT4), NANOG, stage-specific embryonic antigens-3 (SSEA-3), SSEA-4, tumor-related antigen (TRA)-1-60, and TRA-1-81] have been described in MSCs from human fetal but not adult sources (20), raising the possibility that adult MSCs may not possess sufficient plasticity to differentiate towards insulin-producing β-cells. However, several reports demonstrate the expression of these markers in adult stem cells including CB-MSCs (8,10,16,19,26,41,50,52,54,62,64). Here we show stable expression of pluripotent stem cell markers in CB-MSCs through 8–10 passages, and we further demonstrate their downregulation after differentiation.
Recently, the presence of transcription factors (TFs) implicated in β-cell differentiation has been documented in mononuclear cells (MNCs) from CB (45,46). We report here that also CB-MSCs express pancreas-specific TFs prior to differentiation, and we further demonstrate that these TF are stably maintained for at least 5 passages in vitro.
Several studies have demonstrated that CB-MSCs display robust in vitro proliferative capacity without spontaneous differentiation; under appropriate culture conditions, CB-MSCs show the potential to develop into mesodermal-, endodermal-, and ectodermal-derived cell subsets (4,5,28,31,32,35,38,49,61). Although a small number of studies propose that CB-MNCs (13,29,60), “cord blood-derived embryonic-like stem cells” (CBEs) (8,54,63), and CB-MSCs (17,18,24) can differentiate into insulin-producing cells either in vitro or in vivo, there is still the need to carry out a detailed characterization of CB-derived cells to understand their potential clinical use.
Our approach, described herein, is based on the use of stepwise culture protocols where developmental cues are sequentially provided to CB-MSC to induce differentiation towards the pancreatic endocrine phenotype. While similar protocols were initially defined on hESCs (7,34,42), our data are the first that suggest their efficacy on CB-MSCs.
After isolation, expansion and characterization of CB-MSCs from fresh CB samples, we successfully induced CB-MSCs to generate glucose-responsive C-peptide-secreting cell clusters in vitro. Furthermore, we demonstrate glucose-stimulated human C-peptide production in animals that received transplants of partially differentiated CB-MSCs (consisting primarily of pancreatic endodermal cells, with few hormone-expressing cells). Altogether, our results suggest that CB-MSCs have the potential to differentiate into a β-cell-like phenotype, and respond to glucose stimulation both in vitro and in vivo. This is also the first report on pancreatic β-cell differentiation from MSCs following well-established stage-wise protocols.
Materials and Methods
Generation and Expansion of MSCs From Fresh Cord Blood
CB units were received 24–57 h after delivery from the Placenta Blood Program, NY Blood Center, New York, by overnight shipment. Every CB specimen was processed based on unit volume and cell viability, as assessed by trypan blue (0.4%; Invitrogen, Carlsbad, CA) exclusion assay. The MNC fraction from the CB was obtained by Ficoll-Paque PLUS (Stem Cell Technologies Inc., Vancouver, BC, Canada) gradient separation followed by ammonium chloride lysis (Stem Cell Technologies) of red blood cells. Cells were suspended in low-glucose DMEM (Invitrogen) supplemented with 30% fetal bovine serum (FBS), low dexamethasone (10−7 M), penicillin (50 U/ml), streptomycin (50 μg/ml), and l-glutamine (2 mM) (Invitrogen) and plated out at a concentration of 5–7 × 106 cells/ml in T75 culture flasks (BD Biosciences, San Diego, CA). Cells were incubated at 37°C in 5% CO2 in a humidified atmosphere. When well-developed colonies of adherent, fibroblast-like cells appeared, cultures were washed with PBS, harvested with 0.05% trypsin-EDTA (Invitrogen), and reseeded at 1 × 105 cells per flask in the same medium without dexamethasone. When cells reached 80% confluence, they were trypsinized and replated at a 1:3 dilution. Cells expanded from the colonies from each CB unit were stored as different clones in liquid nitrogen for further experiments. For each clone, cells from early passages (p3–p5) were used in all experiments.
To study the doubling/generation time, CB-MSCs (isolated from five different CB units) at passage 4 were seeded in T25 flasks. The cells from one flask were harvested daily and enumerated (three sets of counting/ flask) for 16 consecutive days by the trypan blue exclusion test. The mean of the counts was calculated and plotted against culture time to generate a growth curve. The population doubling (n) level was obtained by the formula: n = log(N/No) × 3.33, where n = number of generations (number of times the cell population doubles during the time interval), N = number of cells at the end of the time interval, and No = number of cells at the beginning of a time interval. The generation time (G) was calculated by the formula: G = t/n (G = generation time; t = time interval in days, hours, or minutes).
Karyotyping and fingerprinting were performed at passages 5 and 35 by cytogenetic standard protocols (Cell Line Genetics, Madison, WI) on one CB-MSC clone (clone #51).
Assessment of Multidifferentiation Potential
CB-MSCs at passage four were cultured in various differentiation media to assess their differentiation potential into adipocytes, osteoblasts, and chondroblasts. MesenCult adipogenic medium (for adipogenesis) and MesenCult Osteogenic Stimulatory Kit (for osteogenesis) (StemCell Technologies) were used and the assays were performed following the manufacturer's protocol. The presence of intracellular lipid droplets in adipocytes was assessed by Oil Red O (Sigma-Aldrich, St. Louis, MO) staining. The mineralization of accumulated calcium, indicative of osteogenic cells, was assessed by Alizarin Red S (Sigma-Aldrich) staining. For chondrogeneis, STEMPRO Chondrogenesis Differentiation Kit (Gibco/ Invitrogen) was used and the assay was performed following the manufacturer's protocol. The presence of proteoglycans, indicative of chondrocytes, was assessed by Alcian Blue (Sigma-Aldrich) staining.
Immunophenotypic Analysis of CB-MSCs by Flow Cytometry
Immunophenotypic analysis of selected cell surface markers was performed on CB-MSCs (n = 7 clones; CB-MSCs isolated from 7 different CB units) at passages 1–6. The following monoclonal antibodies (mAbs) were used for the immunophenotyping of CB-MSCs: fluorescein-isothiocyanate (FITC) conjugated Abs CD3 (S4.1), CD11b (VIM12), CD13 (TÜK1), CD14 (TÜK4), CD29 (MEM-101A), CD31 (MBC78.2), CD34 (581 (class III)), CD44 (MEM-85), CD45 (HI30), CD63 (CLB-gran/12), and CD80 (MEM-233) were from Invitrogen; CD164 (N6B6) and human leukocyte antigen (HLA)-ABC (G46-2.6) were from BD Biosciences. Phycoerythrin (PE) conjugated Ab CD4 (S3.5), CD54 (MEM-111), CD56 (MEM-188), CD103 (LF61), CD105 (SN6) and CD117 (104D2) were from Invitrogen; CD49e (IIA1), CD73 (AD2), CD109 (TEA 2/16), CD122 (Mik-b3), and HLA-DR (G46-6 (L243)) were from BD Biosciences; CD106 (STA), CD144 (16B1), SSEA-3 (eBioMC-631 (MC-631)), SSEA-4 (eBioMC-813-70), TRA 1-60 (TRA 1-60), and TRA 1-81 (TRA 1-81) were from eBioscience (San Diego, CA). Allophycocyanin (APC) conjugated Ab CD8 (3B5), CD10 (MEM-78), CD20 (HI47), CD40 (HB14), and CD86 (BU63) were from Invitrogen; CD28 (CD28.2), CD36 (CB38 (NL07)), and CD81 (JS-81), and CD90 (5E10) were from BD Biosciences; CD133/1 (AC133) was from Miltenyi Biotechnology Inc. (Auburn, CA), and CD184 (12G5) was from eBioscience. Isotype control for each Ab was purchased as suggested from the respective companies.
CB-MSCs were stained with various combinations of mAbs at the recommended concentration by the manufacturer. Cells were detached with 0.05% trypsin-EDTA (Invitrogen), washed twice with fluorescence-activated cell sorting (FACS) buffer (PBS with 2% FBS, 5 mM EDTA and 0.02% sodium azide). A total of 100 μl of cell suspension (~5 × 105 cells) in FACS buffer was aliquoted per tube and appropriately labeled mAbs were added and incubated for 30 min at 4°C in the dark. The cells were then washed twice and resuspended in FACS buffer. Isotype-matched staining with irrelevant Abs was performed to determine background and specificity. Ten thousand labeled cells were used for acquisition, and data were analyzed with CellQuest software on a FACS-Calibur (BD, Becton, Dickinson and Company, Franklin Lakes, NJ).
In Vitro Pancreatic Differentiation of CB-MSCs
Expanded CB-MSCs from early passages (p3–p5) at 80–90% confluence were induced to differentiate towards insulin-secreting cells following protocols published for hESC differentiation by D'Amour et al. (7) and Kroon et al. (34) (Novocell Inc., San Diego, CA). In this study, we refer to CB-MSC differentiation protocols I and II.
CB-MSC differentiation protocol I mimics the five-stage differentiation protocol (S0–S5) originally described by D'Amour et al. (7). The method is as follows. After a brief wash in PBS (with Mg/Ca), the cells were cultured in RPMI (without FBS), activin A (100 ng/ml), and Wnt3a (25 ng/ml) for the first day. The next day, the medium was changed to RPMI with 0.2% v/v FBS and activin A (100 ng/ml), and the cells were cultured for 2 additional days. Next, the cells were briefly washed with PBS (with Mg/Ca) and then cultured in RPMI with 2% v/v FBS, fibroblast growth factor 10 (FGF10; 50 ng/ml), and 3-Keto-N-(aminoethyl-amino-caproyl-dihydrocinnamoyl) (KAAD)-cyclopamine (CYC, 0.25 μM) for 4 days. The medium was then changed to DMEM with 1% v/v B27 supplement, all-trans retinoic acid (RA, 2 μM), CYC (0.25 μM), and FGF10 (50 ng/ ml) and cells were cultured for 4 days. For stage 4, the medium was changed to DMEM with 1% v/v B27 supplement, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT; 1 μM) and exendin-4 (50 ng/ml) and cultured for 3 days. For stage 5, cells were cultured for additional 4 days in CMRL with 1% v/v B27 supplement, exendin-4 (50 ng/ml), insulin-like growth factor (IGF-1; 50 ng/ml), and hepatocyte growth factor (HGF; 50 ng/ml).
The CB-MSC differentiation protocol II comprises the above 5 stages, but with modifications as described previously (34). The first modification eliminates cyclopamine and substitutes keratinocyte growth factor (KGF; 50 ng/ml) for FGF10 during stage 2. The second modification, in stage 3, substitutes Noggin (50 ng/ml) for FGF10. The third modification eliminates DAPT and exendin-4 at stage 4. In addition, to promote transition to definitive endoderm as described for hESCs (42), the PI3K inhibitor LY-294002 (50 μM) was included in CB-MSC differentiation protocol II for the first 3 days of culture during stage 1.
For both protocols, media with appropriate growth factors and supplements were prepared for each stage and sequential, daily changes were made. Media and supplements were obtained from Invitrogen (RPMI, DMEM, B27), R&D Systems (Minneapolis, MN; Wnt3a, activin A, FGF10, KGF, Noggin, IGF-I), Sigma-Aldrich (LY-294002, Exendin4, RA), Toronto Research Chemicals Inc. (Ontario, Canada; KAAD-cyclopamine), and PeproTech Inc., (Rocky Hill, NJ; HGF).
The differentiation process was characterized at each stage by gene expression by real-time polymerase chain reaction (RT-PCR), and protein expression by immunofluorescence (IF) and flow cytometry.
Real-Time Quantitative PCR
Before and after differentiation, cells were harvested and total RNA was isolated using Trizol (Invitrogen) and quantified using an ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). After treatment with DNase I (Invitrogen), cDNA was synthesized by using random primers with the SuperScript-II RT kit following the manufacturer's instructions (Invitrogen). Enough total RNA was converted to cDNA, so that the final concentration was 3–30 ng in 1.5 μl for each 10 μl PCR reaction. Target mRNAs were assayed using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) (Table 1), supplemented with Universal PCR Master Mix (Applied Biosystems). PCR reactions were performed on an Applied Biosystems ABI 7500 real-time PCR thermal cycler.
TaqMan Gene Expression Assays Used for qRT-PCR

Source: All from Applied Biosystems.
Immunofluorescence (IF) Analysis and Confocal Microscopy
Cells were grown in Lab-Tek II chamber slides (Nunc, Rochester, NY) following the differentiation protocol II. At different stages, cells were fixed for 15 min at 24°C in 4% paraformaldehyde in PBS, washed five times in PBS, and blocked for 30 min in PBSTr [0.1% Triton X-100 (Sigma-Aldrich) in PBS] containing 5% normal serum blocking solution (same species as secondary Ab), normal mouse, goat, rabbit serum (Invitrogen), or normal donkey serum (Jackson Immuno Research Laboratories Inc., West Grove, PA) at room temperature. Double IF staining was carried out simultaneously by incubating cells in the mixture of two primary Abs at respective dilutions in blocking buffer in a humidified chamber overnight at 4°C. After decanting and washing three times in PBS, the cells were incubated in the mixture of two secondary Abs (which are raised in different species with two different fluoro-chromes) diluted in blocking buffer in a humidified chamber for 1 h at room temperature in the dark. After decanting and washing in PBS in the dark, the cells were counterstained with DAPI (0.1–1.0 μg/ml) (Santa Cruz Biotechnology, Santa Cruz, CA) for 5 min and rinsed with PBS. For all experiments, control reactions included: 1) the omission of the primary Ab; 2) the replacement of the primary Ab with the appropriate isotype-matched irrelevant Ab; and 3) the omission of the secondary Ab. The stained cells were mounted with a drop of SlowFade Gold antifade reagent (Invitrogen).
The following Abs and dilutions were used: rabbit anti-OCT3/4, 1:500 (SC-9081, Santa Cruz Biotechnology); rat anti-SSEA-3, 1:200 [MC-631/SSEA-3, Developmental Studies Hybridoma Bank (DSHB), University of Iowa, Iowa City, IA]; mouse anti-SSEA-4, 1:200 (MC-813-70/SSEA-4, DSHB); mouse anti-vimentin (VIM), 1:400 (AMF-17-b-c, DSHB); goat anti-pancreatic and duodenal homeobox 1 (PDX1), 10 μg/ml (AF2419, R&D Systems); rabbit anti-neurogenein-3 (NGN3), 1:500 (ab38548, Abcam Inc., Cambridge, MA); rabbit anti-neurogenic differentiation factor 1 (NEUROD1), 5 μg/ml (ab38552, Abcam); mouse anti-NK6 homeobox 1 (NKX6.1), 1:500 (F55A12, DSHB); rabbit anti-islet 1 transcription factor (ISL1), 2 μg/ml (ab20670, Abcam); mouse anti-epithelial cadherin (ECAD), 10 μg/ml (MAB18381, R&D Systems); mouse anti-neuronal cadherin (NCAD), 1:100 (610920, BD Biosciences); goat anti-forkhead box A2 (FOXA2), 10 μg/ml (AF2400, R&D Systems); goat anti-sex determining region Y box 17 (SOX17), 1:200 (SC-17356, Santa Cruz Biotechnology); goat anti-goosecoid homeobox (GSC), 1:200 (SC-22234, Santa Cruz Biotechnology); mouse anti-v-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), 10 μg/ml (ab57807, Abcam); rabbit anti-MAFB, 10 μg/ml (ab56242, Abcam); rabbit anti-pancreatic polypeptide (PPY), 1:200 (AB939, Chemicon, Billerica, MA); rabbit anti-glucagon(GCG), 1:500 (PU039-UP, Biogenex, San Ramon, CA); mouse anti-C-peptide/proinsulin, 1:500 (CBL94, Chemicon, note that this Ab detects both C-peptide and proinsulin); guinea pig anti-insulin (INS), 1:500 (A0564, Dako North America, Inc., Carpinteria, CA). Alexa-488-, Alexa-555-, and Alexa-568-conjugated goat and donkey Abs against mouse, rat, rabbit, guinea pig, and goat (Molecular Probes/ Invitrogen) were used at 1:500 dilutions.
For fluorescence imaging, a Leica DM IRB microscope (Leica Microsystems, Bannockburn, IL) was used, and the images were acquired digitally using a high-resolution B/W CCD digital camera ORCA-ER (Hamamatsu Corp., Bridgewater, NJ). Fluorescence images were also acquired, as needed, on a Zeiss LSM510 confocal microscope (Carl Zeiss Inc., Thornwood, NY). Quantitative analysis was carried out by counting the number of immunoreactive cells and comparing that number to the total number of viable cells as determined by DAPI staining. A minimum of 20 random fields were counted for each marker.
Glucose-Stimulated C-Peptide Release In Vitro
Glucose-stimulated C-peptide release was assessed as described by Chen et al. (6) by incubating the differentiated CB-MSCs in one well of a six-well plate with Krebs-Ringer solution containing bicarbonate and HEPES [KRBH; 129 mM NaCl, 4.8 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 5 mM NaHCO3, 10 mM HEPES, 0.1% (w/v) BSA] (Sigma-Aldrich). The cells were incubated with KRBH buffer for 1 h as washing. The cells were incubated with KRBH buffer with 2.5 mM d-glucose for 1 h and then KRBH buffer with 20 mM d-glucose for 1 h. The C-peptide levels in culture supernatants were measured using the Human Ultrasensitive C-peptide enzyme-linked immunosorbent assay (ELISA) kit (Mercodia, Uppsala, Sweden) following the manufacturer's instructions, and normalized to total cellular DNA. Genomic DNA from cells was isolated using PureLink Genomic DNA Mini Kit (Invitrogen) and quantified using ND-1000 spectrophotometer (NanoDrop).
In Vivo Experiments
NOD.CB17-Prkdcscid/J [nonobese diabetic severe combined immunodeficient (NOD-SCID)] mice (7–8 weeks old, male) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in virus antibody-free rooms in microisolator cages with free access to sterilized food and water. All animal manipulations were conducted at the Preclinical Cell Processing and Translational Models Core of the Diabetes Research Institute under protocols reviewed and approved by the University of Miami's Institutional Animal Care and Use Committee.
Partially differentiated CB-MSCs in culture to the pancreatic endoderm stage (stage 4 at day 14 from the second differentiation protocol) were used for transplantation procedures. The cell aggregates were collected by gentle scraping, allowed to settle by gravity and kept on ice in RPMI containing B-27 serum-free supplement. Slurry of 10–25 μl of cell aggregates (5–7 × 106 cells) in saline solution was delivered either by intraperitoneal (IP) injection, or transplanted under the kidney capsule. Briefly, under general anesthesia, the left kidney was exposed and a small opening was made in the kidney capsule. Cells were delivered under the kidney capsule with a PE-50 catheter. The kidney was repositioned and the muscular and skin layers sutured. In the sham control group, a cell-free saline injection was made via identical procedures.
Glucose-Stimulated C-Peptide Secretion in Transplanted Mice
For the detection of human C-peptide, animals were fasted overnight (~18 h), and an IP bolus of glucose was administered (2.0 g/kg body weight) at selected time points during the follow-up period. Blood samples were obtained 1 h after the bolus from the retro-orbital plexus under general anesthesia. Human C-peptide levels in plasma were measured with the Ultrasensitive Human C-peptide ELISA kit (Mercodia) according to the manufacturer's instructions.
Statistical Analysis
Results are expressed as mean ± SD. Statistical significance of differences was assessed by the Student's t-test. A value of p < 0.05 was considered statistically significant.
Results
Isolation, Expansion, and Growth Characteristics of CB-MSCs
Out of ~250 CB units, 90 were selected based on CB volume (>80 ml; range 40–130 ml) and cell viability (>70% viability; range 50–90%) and processed in an attempt to generate MSC colonies. Colonies were successfully generated from 31 out of the 90 CB units processed (frequency 34.4%). No correlation was detected between hours elapsed after CB collection, volume, number of MNCs in the CB, and the success in the generation of MSCs. At 8–25 days (mean 14) after seeding of MNCs, adherent cells of fibroblast-like morphology were detected with a median frequency of five colonies per CB (range 1–18). After the first passage, highly proliferating fibroblast-like cells became predominant over other cell types. Over the following two passages, adherent cells were almost entirely composed of spindle-shaped, fibroblast-like cells that later grew to confluence in 8–12 days (Fig. 1A). CB-MSC populations from individual CB units exhibited differences in growth dynamics, from slow growing clones to very rapidly growing ones. Growth curve studies indicated that the cells doubled every ~4.6 ± 2.8 days (range 1.6–8.6 days) (Fig. 1B). Some CB-MSC clones slowed down proliferation after passage 7 or 8, whereas other clones could be expanded every 2–3 days for 20–30 passages and beyond. During the cultures, the nucleus/cytoplasm (N/ C) ratio decreased, suggesting that the cells entered senescence (data not shown).
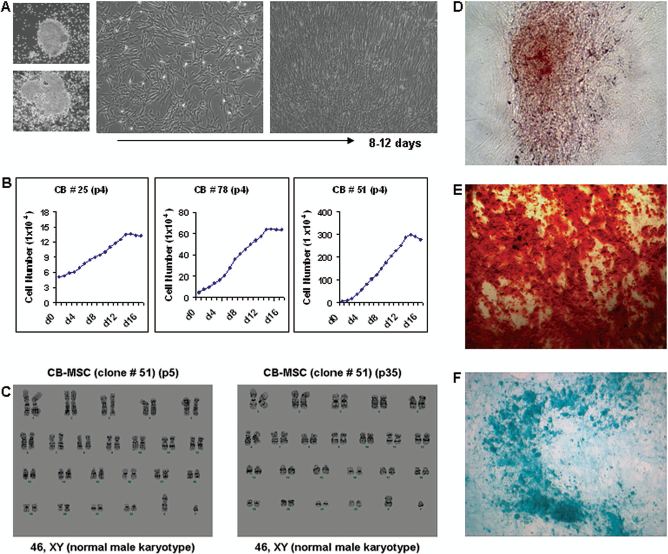
Characteristics of cord blood-derived mesenchymal stem cells (CB-MSCs). (A) Adherently growing colonies from CB-mononuclear cells (MNCs) expand to spindle-shaped fibroblast-like MSCs (original magnification: 20×). (B) Expansion kinetics of CB-MSCs plated at low densities. The growth rates of CB-MSCs varied from slow growing to very rapidly growing clones. (C) Karyotype analysis of CB-MSCs. Cytogenetic analysis was performed for CB-MSCs (clone #51) in early and late passages (p5 and p35) to determine whether the polyploidy seen in prolonged cell culture resulted in altered chromosome content. All 20 cells in both passages maintained the normal 46XY karyotype and no abnormal cells were detected. (D–F) CB-MSCs undergo in vitro differentiation into adipogenic, osteogenic, and chondrogenic lineages (original magnification: 20×).
Cytogenetic analysis was performed on 20 G-banded metaphase cells from CB-MSCs derived from one clone (CB-51) at passage 5 and passage 35. All cells in both passages demonstrated an apparently normal 46XY karyotype, and no gross abnormalities could be detected (Fig. 1C). CB-MSCs cultured in appropriate differentiation media showed adipogenic, osteogenic, or chondrogenic differentiation potential (Fig. 1D–F).
CB-MSCs Express the Characteristic MSC Markers
Phenotypic analyses were performed on different CB-MSC clones cultured from passages 2 through 6 by flow cytometry. The CB-MSCs (n = 6, passage 4) expressed most of the typical MSC surface markers. The frequency percentage was high for CD10 (93.2 ± 3.6%), CD13 (93.1 ± 5.1%), CD29 (97.1 ± 1.5%), CD44 (98.8 ± 1.1%), CD49e (97.4 ± 0.9%), CD73 (97.9 ± 1.7%), CD81 (97.4 ± 2.3%), CD90 (98.1 ± 0.6%), CD105 (97.1 ± 1.5%), HLA-ABC (96.1 ± 2.1%) and variable for CD54 (25.1 = 79.6%), CD56 (20.2 = 32.5%), CD63 (27.73 = 98.5%), and CD106 (35.4 = 92.5%) (Fig. 2A). CB-MSCs were negative for macrophage markers (CD11b and CD14), costimulatory molecules (CD28, CD40, CD80, and CD86), hematopoietic cell markers (CD34, CD45, CD117, and CD133), adhesion molecules (CD31/PECAM1), endothelial markers (CD36 and CD144), integrin, alpha E (CD103), and major histocompatibility complex (MHC) class II (HLA-DR) (Fig. 2B). They were also negative for CD3, CD4, and CD8 (T-cell markers), CD20 (B-cell marker), CD164 (adhesion molecule), and CD184/CXCR4 (endothelial cell marker/hematopoietic cell marker) (data not shown).

Immunophenotype of CB-MSCs. Cells were labeled with the mAb specific for the molecule indicated (filled histograms) or isotype controls (open histograms). CD, cluster of differentiation; HLA, human leukocyte antigen.
CB-MSCs Express Pluripotent Stem Cell Markers
Several recent publications have demonstrated that, like hESCs, adult stem cells may also occasionally express pluripotent markers such as NANOG, OCT4, SSEA-3, SSEA-4, TRA-1-60, and TRA-1-81 (8,10,16,19,26,41,50,52,54,62,64). In order to determine whether this was also the case in CB-MSCs in our study, we performed additional IF analyses on undifferentiated cells. OCT4 was found to be highly expressed in the nucleus of ~80–90% of the cells (Fig. 3A). Staining for the membrane markers of pluripotency SSEA-3 and SSEA-4 was also positive in ~30–40% of the cells (Fig. 3A). Expression of TRA-1-60 and TRA-1-81, in contrast, was very weak or undetectable by flow cytometry (data not shown).

Immunofluorescence analysis of undifferentiated CB-MSCs. (A) Expanded CB-MSCs express the pluripotent stem cell markers octamer-binding transcription factor 4 (OCT4), stage-specific embryonic antigen-3 (SSEA-3) and SSEA-4 and (B) pancreatic transcription factors (TFs)/precursor markers [pancreatic and duodenal homeobox 1 (PDX1), neurogenin-3 (NGN3), neurogenic differentiation factor 1 (NEUROD1), NK6 homeobox 1 (NKX6.1), islet 1 transcription factor (ISL1)], and the mesenchymal marker vimentin (VIM). Coexpression of selected markers is shown in (C). DAPI (blue) labels cell nuclei. Images were taken from CB-MSCs (clone #51, p3) using a Leica DM IRB inverted microscope (A, C) and an LSM510 confocal microscope (B) (original magnification: 40x). Similar staining results were obtained in three different clones at both passages (p3 and p7) tested.
Expanded, Undifferentiated CB-MSCs Express Markers of Pancreatic Endocrine Cells
Two recent reports demonstrate that freshly isolated and expanded CB-MNCs express some markers typically associated with pancreatic development (45,46). To determine if CB-MSCs also comprised such “pancreas-committed” subpopulations, immunofluorescence staining for pancreatic endocrine cell markers was carried out in three different CB-MSC clones at early passage levels (p3 and p7). Indeed, we found strong nuclear staining for the key pancreatic TFs in a relatively large proportion (30–40% for PDX1, NKX6.1, and ISL1; 70–80% for NGN3 and NEUROD1) of CB-MSCs in all three clones tested (Fig. 3B). They also stained positive for the MSC cell membrane marker vimentin (Fig. 3B). Staining for FOXA2 and SOX17 was negative (data not shown). Coexpression of vimentin with NGN3, NEUROD1, and ISL1 was observed in ~60–70% of the triple stained cells (Fig. 3C).
Directed Differentiation of CB-MSCs
Undifferentiated CB-MSCs underwent considerable morphological changes during the various stages of differentiation of protocol I (Fig. 4A, B). The effects of various treatments on differentiation were initially evaluated using real-time PCR to detect gene expression characteristic of each cell population at different stages. CB-MSC to mesendodermal (ME) cell transition from stage 0 to stage 1 was associated with downregulation of the pluripotent stem cell marker OCT4 and the epithelial-type cadherin (ECAD). At the same time, there was upregulation of definitive endoderm (DE) markers [chemokine (C-X-C motif) receptor 4 (CXCR4) and SOX17], posterior foregut endoderm (PF) markers [PDX1 and motor neuron and pancreas homeobox 1 (HLXB9)], pancreatic endoderm (PE) markers (NGN3 and NKX6.1), and the immature α/β-cell marker paired box 6 (PAX6). However, upregulation of most of these markers, with the exception of HLXB9 and NKX6.1 (40–50-fold), was relatively small (5–12-fold up). The primitive gut tube (PG) markers hepatocyte nuclear factor 1B (HNF1B) and HNF4A, PE markers PAX4 and MAFB, and the endocrine cell markers INS, GCG, and PPY could not be detected (Fig. 4C).

CB-MSC differentiation to hormone expressing endocrine cell (protocol I). (A) Schematic representation of the five-stage differentiation protocol I [adapted from D'Amour et al. (7)] for CB-MSCs through mesendoderm (ME) to definitive endoderm (DE), primitive gut tube (PG), posterior foregut (PF), pancreatic endoderm (PE), and, finally, hormone-expressing endocrine cells (EN) with applied factors and media shown here and further explained in Materials and Methods. Several markers characteristic of each differentiation stage/cell population are listed. ActA, activinA; Wnt, Wnt3; FGF, fibroblast growth factor; CYC, KAAD-cyclopamine; RA, all-trans retinoic acid; DAPT, γ-secretase inhibitor; Ex4, exendin-4; IGF, insulin-like growth factor; HGF, hepatocyte growth factor. (B) Changes in cell morphology and cluster formation during in vitro differentiation of CB-MSCs. (C) Temporal expression of pancreatic lineage genes during CB-MSC-derived islet-like cluster (ILC) generation. CB-MSCs (n = 5) were differentiated to ILCs following the differentiation protocol I. Cell samples were collected for each stage at days 1, 4, 8, 12, 15, and 19 and analyzed by qRT-PCR for OCT4, ECAD, SOX17, FOXA2, CXCR4, HNF1B, HNF4A, PDX1, HLXB9, NGN3, PAX4, NKX6.1, PAX6, insulin, and glucagon expression (see Materials and Methods for definitions). All mRNA expression levels were normalized to the housekeeping gene 18S rRNA expression. Relative gene expression (RQ) was determined by normalization to that of undifferentiated CB-MSC sample at stage 0 (S0) in the dataset and the standard deviation of the gene expression measurements was reported.
Figure 5 looks at the second differentiation protocol (Fig. 5A), which resulted in the development of well-defined cell clusters at stage 5 (Fig. 5B). The decrease in the expression of OCT4 and ECAD in stage 1 was more marked, to the point that these markers were nearly undetectable at stage 3. IF analyses also showed absence of ECAD expression, and upregulation of neural-type cadherin (NCAD) at stage 1, suggesting the transition of CB-MSCs to ME cells (Fig. 6A, B). Expression of NCAD declined in stage 2 and was not observed in later stages (Fig. 6A). Expression of the DE markers SOX1 and CXCR4 was observed throughout stages 1 and 2, gradually declining from stage 3 onwards (Figs. 5C and 6C). The DE marker FOXA2 was not seen either at mRNA or protein levels. Positive IF staining for the ME/PE marker goosecoid (GSC) was also observed in stage 1 (data not shown). In stage 2, we observed the expression of the primitive gut marker HNF4A, but not HNF1B. In stage 3, the cells expressed high levels of PDX1 and HLXB9 while maintaining the expression of HNF4A (Fig. 5C), suggesting transition towards PG. The pancreatic precursor/PG/PE markers PDX1, NGN3, NEUROD1, and NKX6.1 were expressed at the mRNA and protein levels in stages 3 and 4 (Figs. 5C, 6D–F), reflecting the initial commitment of these cells to the endocrine lineage. In stage 4, the PE marker PAX4 could not be detected.

CB-MSC differentiation to hormone-expressing endocrine cell (protocol II). (A) Schematic representation of the five-stage differentiation protocol II [adapted from Kroon et al. (34) for stages 1–4 and from D'Amour et al. (7) for stage 5] for CB-MSCs through mesendoderm (ME) to definitive endoderm (DE), primitive gut tube (PG), posterior foregut (PF), pancreatic endoderm (PE), and finally hormone-expressing endocrine cells (EN) with applied factors and media shown here and further explained in Materials and Methods. Several markers characteristic of each cell population are listed. LY, LY-294002; ActA, activinA; Wnt, Wnt3; KGF, keratinocyte growth factor; RA, all-trans retinoic acid; Cyc, KAAD-cyclopamine; Nog, noggin; Ex4, exendin-4; IGF, insulin-like growth factor; HGF, hepatocyte growth factor. (B) Changes in cell morphology and cluster formation during in vitro differentiation of CB-MSCs. (C) Temporal expression of pancreatic lineage genes during CB-MSC-derived islet-like cluster (ILC) generation. CB-MSCs (n = 5) were differentiated to ILCs following the differentiation protocol II. Cell samples were collected for each stage at days 1, 4, 7, 10, 13, and 17 and analyzed by qRT-PCR for OCT4, ECAD, SOX17, FOXA2, CXCR4, HNF1B, HNF4A, PDX1, HLXB9, NGN3, PAX4, NKX6.1, PAX6, insulin, and glucagon expression. All mRNA expression levels were normalized to the housekeeping gene 18S rRNA expression. Relative gene expression (RQ) was determined by normalization to that in undifferentiated CB-MSC sample at stage 0 (S0) in the dataset and the standard deviation of the gene expression measurements was reported. (D) Flow cytometric staining for the expression of pluripotent stem cell markers, SSEA-3 and SSEA-4 at stage 0 and stage 5 in differentiating CB-MSCs (n = 5). The expression of both these markers was not detected (0%) in CB-MSCs at stage 5, indicating that these cells lose their pluripotency as they progress with the differentiation.
The appearance of the first endocrine cells at stage 5 was marked by the expression of the TFs MAFA and PAX6 by RT-PCR (Fig. 5C). PDX1 and NGN3 expression remained constant at stage 5 (Fig. 5C). PDX1, NKX6.1, and the endocrine progenitor markers NEUROD1 and ISL1 were detected at the protein level in stage 5 (Fig. 7A). PDX1, NGN3, and NEUROD1 expression at the protein level (40–50%) was observed in all stages (S0–S5, data not shown). MAFB expression was observed in stage 4 but declined in stage 5, whereas MAFA expression was observed only in stage 5 at both mRNA and protein levels (Figs. 5C and 7B), possibly indicating the differentiation and progressive maturation of insulin-producing cells. Staining for SSEA-3 and SSEA-4 (which was positive in CB-MSCs prior to differentiation) markers was completely negative (SSEA-3: down from ~15–25% to 0% and SSEA-4: down from ~25–35% to 0%) at this stage, as shown in Figure 5D.

Immunofluorescence analysis of transitions from definitive endoderm to pancreatic endoderm and endocrine precursor cells in differentiating CB-MSCs (clone #51). The CB-MSC differentiation protocol II was followed as described in Materials and Methods. (A, B) CB-MSC to mesoenddermal (ME) cell transition is associated with the downregulation of the epithelial-type cadherin (ECAD, green) from stage 0 to stage 2 and the upregulation of neural-type cadherin (NCAD, green) in the newly formed ME cells at stage 1. NCAD expression declined in stage 2 and was not observed in further stages (S3–S5). (C) Fluorescence micrographs showing the expression of definitive endoderm (DE) marker SOX17 (red) at stages 1 and 2, which continued to express in some cells at stage 3. (D–F) Staining for PDX1 (green), NGN3 (red), and NEUROD1 (green) at stage 3 [transition from DE to posterior foregut (PF)] and at stage 4 ([transition from PF to pancreatic endoderm (PE)]. DAPI (blue) labels cell nuclei. Images were taken using a Leica DM IRB inverted microscope (original magnification: 20×). Similar results were obtained in all 3 CB-MSC clones used for differentiation experiments.

Immunofluorescence analysis of insulin gene transcription factors and MAF factors during the differentiation of pancreatic endocrine cells from CB-MSCs (clone #51). The CB-MSC differentiation protocol II was followed as described in Materials and Methods. (A) The expression of pancreatic endoderm markers (PDX1, NKX6.1) and endocrine progenitor markers (NEUROD1, ISL1) was observed at the protein level in stage 5. (B) Differentiated CB-MSCs expressed MAFB (red) in stage 4 and was detected faintly in some cells at stage 5. MAFA (green)-expressing cells were detected only at the last stage (stage 5), representing the endocrine precursor cell transition from stage 4 into insulin-producing cells. DAPI (blue) labels cell nuclei. Images were taken using Leica DM IRB inverted microscope (original magnification: 20×). Similar results were obtained in all 3 CB-MSC clones used for differentiation experiments.
Differentiated CB-MSCs Produce Endocrine Pancreatic Hormones
Endocrine cells that express the pancreatic hormones insulin (INS), glucagon (GCG), and pancreatic polypeptide (PPY) are generated throughout stage 5 (about 16–18 days of CB-MSC differentiation) (Figs. 5C and 8A–C). As determined by IF, the number of PPY-expressing cells was relatively high compared to that of GCG-expressing cells (Fig. 8A). A smaller proportion of endocrine cells derived from CB-MSCs coexpressed INS and GCG or INS and PPY (data not shown), as reported in hESC-derived endocrine cells (7).

Immunofluorescence analysis of hormone-expressing cells in differentiating CB-MSCs (clone #51). The CB-MSC differentiation protocol II was followed as described in Materials and Methods. (A) End stage CB-MSCs were positive for the pancreatic hormones pancreatic polypeptide (PPY, red) and glucagon (GCG, red). (B, C) Triple staining showing the expression of insulin (INS, green) and C-peptide (C-PEP, red) and coexpression (INS/C-PEP, yellow) in islet-like clusters (ILCs) derived from CB-MSCs at the final stage of differentiation. In these ILCs, cells were aggregated into different size clusters and the cells appeared smaller in big clusters than in small clusters. In big ILCs, specific labeling of INS is evidenced by the presence of “ring-like” stain (green arrow) at the rim of the big cluster (B). DAPI (blue) labels cell nuclei. Images were taken using a Leica DM IRB inverted microscope (for A and B, original magnification: 20×). To visualize colocalization of INS and C-PEP in small clusters, images were taken at higher magnification using a Zeiss LSM510 confocal microscope (C, original magnification: 40x). Similar results were obtained in all three CB-MSC clones used for differentiation experiments.
As shown in Figure 5B, islet-like cell clusters (ILCs) were developed from CB-MSCs at the final stage of differentiation. As the endocrine cells aggregated into clusters of different sizes, the cells in large clusters appeared smaller than those in small ones. IF staining of ILCs showed the expression of INS and C-peptide (Fig. 8B, C). The expression of C-peptide in these clusters suggests that proinsulin was newly synthesized during the later stages of differentiation (the C-peptide Ab used in this study detects both C-peptide and proinsulin). To analyze coexpression of INS and C-peptide, specific areas of the culture were imaged where endocrine cells in small clusters were present primarily in monolayers (Fig. 8B, C).
Glucose-Induced C-Peptide Release by CB-MSC-Derived Endocrine Cells
Glucose sensing and insulin secretion are hallmarks of pancreatic β-cell function. To confirm the de novo synthesis and release of insulin by CB-MSC-derived insulin-expressing cells, we monitored the release of C-peptide into the culture medium in response to glucose stimulation. Exposure of cells to 20 mM glucose increased the C-peptide release approximately fourfold over basal (2.5 mM) glucose stimulation (9.02 ± 1.37 vs. 2.13 ± 0.45 pmol/μg DNA, p < 0.0001) (Fig. 9A), in a manner comparable to that of adult human islets (60.00 ± 4.54 vs. 14.95 ± 2.00 pmol/μg DNA, p < 0.0001).

In vivo tissue distribution of CB-MSCs and C-peptide release. (A) Glucose-stimulated C-peptide release by differentiated CB-MSCs in vitro. C-peptide secretion assay was performed by static incubation and measured by ELISA as described in Materials and Methods and normalized to DNA content. C-peptide level in the 20 mM glucose medium was fourfold higher than that in the 2.5 mM glucose medium (*p < 0.0001). Results are the means of four experiments with different CB-MSC clones. (B-a) Detection of CB-MSCs in transplanted mice. Mice were sacrificed 1 week after transplantation and single-cell suspensions were obtained from bone marrow, spleen, thymus, lymph nodes, and blood and analyzed by flow cytometry. (B-b) Except the control group, analysis of blood from experimental groups—mice transplanted under the kidney capsule in both kidneys (2 KdCp) or in one kidney (1 KdCp) or injected intraperitoneally (IP)—revealed the presence of HLA-ABC+ and CD81+ cells (frequency ~3–6%) specific for the transplanted CB-MSCs. (C) Plasma C-peptide levels in transplanted mice. Mice transplanted with CB-MSC-derived pancreatic endocrine (PE) cells or human islets were analyzed at the postengraftment times (day 30, 60, and 150) for the plasma levels of human C-peptide as described in Materials and Methods. C-peptide was not detected in the control group (mice without transplant, n = 3) while a transient increase (~5–20-fold) in the serum levels of human C-peptide was observed in experimental mice (CB-MSC transplantation under kidney capsule, n = 12; by IP injection, n = 2) at both day 60 (p < 0.0885) and day 150 (*p < 0.045) time points. The Ultrasensitive Human C-Peptide ELISA kit (Mercodia) used in this assay has no cross-reactivity to mouse C-peptide and has a detection limit of 1.5 pmol/ml. The reference levels of human C-peptide in normoglycemic nude diabetic mice transplanted with human islets were ~55 pmol/ml before (at fasting) and ~200 pmol/ml at 60 min after glucose stimulation (not shown).
In Vivo Differentiation of Engrafted CB-MSC-Derived Pancreatic Endodermal Cells Into Functional Endocrine Cells
To provide evidence that CB-MSC-derived pancreatic endoderm can generate functional endocrine cells in vivo, we transplanted pancreatic endoderm stage (stage 4) cell aggregates collected from the second differentiation protocol into immunodeficient (NOD/SCID) mice either IP or under the kidney capsule. Recipient animals were humanely euthanized at different time points after the procedure, and single-cell suspensions were obtained from spleen, lymph nodes (cervical, mesenteric, and iliac), thymus, bone marrow, and blood. These samples were stained and analyzed for the presence of HLA-ABC+ and CD81+ CB-MSCs by flow cytometry. At 7 days posttransplantation, we were able to detect donor cells (~3–6%) in the blood (Fig. 9B-a, B-b) and bone marrow (data not shown) but not in the spleen, thymus, or lymph nodes. However, we were unable to track these cells at later time points (day 30 and 60). Notably, no teratoma formation was observed in CB-MSC recipients at any of the time points of our study.
Starting at day 30 after implantation, we assessed glucose-stimulated C-peptide secretion in plasma collected from recipient mice. While no human C-peptide could be demonstrated 30 days posttransplantation, low levels of the protein were detectable around day 60, after a glucose challenge. Measurable levels of human C-peptide were again detected in response to glucose administration at day 150 posttransplantation (range 0.0–7.97 pmol/ml). This ~5–20-fold increase (transplantation under kidney capsule, n = 12; by IP injection, n = 2) at day 150 posttransplant was statistically significant (p < 0.045) (Fig. 9C). Human C-peptide was always undetectable in the control group (mice without transplant, n = 3).
Discussion
Human umbilical cord blood cells are a readily available potential source of insulin-producing cells for transplantation. They are in plentiful supply and can be banked for a wide HLA representation or prospectively stored from individuals at birth for future autologous transplantation. Unlike other MSCs, those from the cord blood arise from the youngest possible postnatal tissue, and as such are expected to retain, at least in theory, higher developmental plasticity and potential for replication. Since their procurement is not controversial, they could have an edge over hES cells provided that they could be instructed to differentiate into insulin-producing β-like cells. Detractors of MSC-based approaches, in contrast, argue that these cells are already mesodermal and therefore cannot be effectively pushed along the true endodermal β-cell lineage.
The CB-MSCs herein described satisfy all three criteria set forth by the International Society for Cell Therapy to define MSCs, namely (i) plastic adherence of the isolated cells in culture and (ii) expression of CD29, CD44, CD73, CD90, and CD105 in greater than 95% of the culture, and lack of expression of markers such as CD34, CD45. and HLA-DR and (iii) the differentiation potential into fat, bone, and cartilage (12). Our results confirm other observations (5,28,31,35) that cells with similar cell surface antigen profile can differentiate into bone, fat, and cartilage, and strongly suggest that they are indeed MSCs. MSCs typically have a limited life span and ultimately undergo replicative senescence, manifested by loss of proliferation and altered morphology. Analysis of senescence in our individual colonies and/or CB units suggests remarkable variability, confirming observations reported earlier by Kern et al. (28). Some of our CB-MSC clones expanded up to 20–40 passages for >6 months without visible changes in either growth pattern or morphology. Although the MSCs described here can be cultured and expanded from a relatively low number of CB units (~35%), they can be maintained with a normal karyotype for several passages (up to 35).
Archetypal stem cell markers such as NANOG, OCT4, stage-specific embryonic antigens (SSEA-3 and SSEA-4) and tumor rejection antigens (TRA-1-60 and TRA-1-81) were previously thought to be specific to very early embryonic development and to hESCs. However, expression of these markers in adult stem cells has been documented recently (8,10,16,19,26,41,50,52,54,62,64). OCT4 is considered a master regulator of hESC pluripotency and self-renewal capacities. “Stemness” properties are attributed to OCT4A, one of the two spliced variants of OCT4 gene. The function of the other variant, OCT4B, remains unknown (2,33). While the expression pattern and function of OCT4A in somatic stem cells remain controversial (36,37), Seo et al. demonstrated recently that inhibiting OCT4 in CB-MSCs resulted in decreased cell proliferation and reduced multipotency (53). Expanding upon such findings, we show that CB-MSCs express ECAD, SSEA-3, and SSEA-4 in addition to OCT4 (the OCT4 antibody used in our study is non-cross-reactive with OCT4 isoform B). This observation led us to hypothesize that these CB-MSCs might not be fully committed to the mesodermal fate, as is the case with a majority of mesenchymal cell types. Such well-documented commitment has been presented as a major roadblock for the design of protocols aimed at generating of endoderm-derived tissues, such as pancreatic β-cells. In most instances, such efforts have led to the inefficient induction of mixed phenotypes inconsistent with what we have come to expect of bona fide, fully functional β-cells. Our finding that CB-MSCs respond to a chemical β-cell differentiation protocol originally devised for pluripotent cells (7,34,42) is novel and indicates that, while sharing key properties and markers with MSCs, these cells are more primitive. The downregulation of “stemness” genes as differentiation progresses is suggestive that their expression might be associated to the multipotency of CB-MSCs.
Expression of TFs implicated in islet development such as ISL1, NKX2.2, NKX6.1, NGN3, PDX1, and NEUROD1 has been reported in undifferentiated, human exocrine pancreas-derived MSCs (3). At variance, expanded MSCs derived from human bone marrow were shown to express NKX6.1 constitutively but none of the other TFs of the β-cell developmental pathway (44). In vitro-proliferating human adipose tissue-derived MSCs were shown to express ISL1, which is a crucial TF for the development of pancreatic endocrine cells (56). The expression of selected key pancreas-specific TFs has also been recently reported in freshly isolated CB-MNCs and uninduced/expanded CB-MNCs at early passages (45,46). The first report (46) suggests that the strongly adherent cells grown from CB-MNCs morphologically resemble the MSCs derived from human bone marrow and demonstrate the presence of cells expressing NGN3, ISL1, and PAX4, but not PDX1. However, there are no immunophenotypical data in this report to unequivocally establish that these CB-MNC-derived adherent cells were MSCs. Similarly, the second report (45) described that freshly isolated CB-MNCs, as well as MSC-like cells derived from CB-MNCs in vitro, express the key pancreatic TFs PDX1, NGN3, ISL1, brain-4 (BRN4), and PAX6. These cells were not pure MSCs either, as they had markers for both MSCs (CD44: 30–36%; CD90: 1–13%) and hematopoietic cells (CD45: 45%).
Here we report, for the first time, that expanded populations of undifferentiated CB-MSCs express the key pancreatic factors PDX1, NGN3, NEUROD1, NKX6.1, and ISL1 in a relatively large proportion of CB-MSCs. Coexpression of vimentin with NGN3, NEUROD1, and ISL1 was observed in ~60–70% of the triple stained cells, indicating the presence of a subpopulation of cells within the undifferentiated CB-MSCs that may be pre-committed towards the pancreatic lineage. Of note, many of these pancreatic markers are also typically found during neural development (9,11). Therefore, it is important to stress that the mere presence of such markers would not be indicative per se of pancreatic commitment. However, the subsequent differentiation of CB-MSCs into insulin-positive, glucose-responsive cells is consistent with the above hypothesis.
Mimicking natural pancreatic development is arguably the best strategy to produce glucose-responsive, insulin-producing cells in vitro. As previously described for hESCs (7,34), we have established that this approach can be used to differentiate CB-MSCs in vitro through stages that resemble definitive endoderm, pancreatic endoderm, and endocrine precursors towards cells that produce islet hormones such as insulin, C-peptide, glucagon, and pancreatic polypeptide. Similar to the results shown for hESCs by Mc Lean et al. (42), we also demonstrate that inhibition of PI3K signaling efficiently promotes differentiation of CB-MSCs into mesendoderm and then definitive endoderm. The differentiation process occurred in a stage-wise fashion, showing a sequential pattern of expression consistent with that observed during pancreatic development. Unlike previous reports on hESCs (7), our CB-MSC-derived cells made the switch from MAFB+MAFA- to MAFB-MAFA+ while maintaining PDX1 and NKX6.1, clearly indicating the maturation of insulin-producing cells in stage 5.
During stage 5 (day ~15–18), cells expressing at least three out of the five pancreatic endocrine hormones (C-PEP, INS, GCG, and PPY) could be detected. As observed by others (7,48), a small number of CB-MSC-derived endocrine cells express more than one hormone. Though D'Amour et al. observed a robust increase in C-peptide release to different in vitro responses, the C-peptide secretion in their cultures was only minimally responsive to glucose (approximately twofold increase to basal level), similar to that of immature fetal β-cells (7). Our cultures exhibit a stronger response to glucose challenge in vitro.
Previous studies have shown in vivo maturation of hESC-derived pancreatic precursors obtained using the same differentiation protocols utilized in our study and measured as a progressive increase of human C-peptide secretion (both fasting and stimulated) in recipient mice (34). In our study, transplantation of CB-MSC-derived pancreatic endocrine cell precursors indeed led to measurable C-peptide secretion, but the C-peptide levels were significantly lower than those previously described for hESC-derived pancreatic precursors generated using similar protocols (34). Therefore, it is unlikely that the levels measured in our study could sustain normoglycemia in diabetic mice. This clearly points to the necessity of qualitatively/quantitatively improving the mass of glucose-responsive, insulin-producing cells before this approach can be considered for clinical transplantation.
We can only speculate about the differential response observed in CB-MSCs versus hESCs subjected to the same treatment. In all likelihood these differences are just a natural consequence of the diverse nature of the starting tissue. Despite the primitiveness of these CB-MSCs, one of our most relevant findings is that they express high levels of pancreatic markers such as NGN3 or NKX6.1 even before they have been induced to differentiate. From this perspective, a lower-than-expected relative upregulation of these genes may simply reflect that these cells were not at the same developmental stage at the initiation of the protocol. However, we cannot assume either that CB-MSCs are equivalent to hES cells at stage 4 of differentiation, because MSCs are not endodermal in origin. In fact, in vitro evidence is insufficient to indicate that these cells are reproducing any native ontogenic/regeneration process, as it is well known that (a) cultures are a poor representation of the in vivo environment where these phenomena take place and (b) adaptation of MSCs to growth in plastic is associated with epigenetic changes that invariably distort their “normal” physiological responses anyway. Whether or not this process recapitulates a natural one, the fact remains that the differentiation of CB-MSCs into glucose-responsive, insulin-secreting cells has great potential for the development of cell therapies for diabetes.
Recent studies demonstrated that autologous transfusion of unmanipulated umbilical CB in subjects with type 1 diabetes at onset is safe (22,23) and potentially beneficial to slow disease progression. These trials were based on the hypothesis that CB cells may improve outcomes through immunomodulation by selected cell subsets comprising MSCs. The “immunosuppressive” property of human MSCs makes them an important candidate for cellular therapy also in allogeneic settings (30,58). CB-stem cells may therefore represent a source of cell precursors with multiple functions, and approaches encompassing regenerative medicine and immunomodulation could be envisioned.
The use of CB-MSCs may also offer additional advantages, when compared to hESCs, which include the large availability and ease of procurement, and the possibility of creating large banks that may match most or all prospective recipients. Unlike hESCs that are prone to the development of teratomas in vivo (6,14,34), no teratomas were observed after transplantation of human CB-MSCs in immunodeficient recipients in our study. This observation, along with the stability of the CB-MSC phenotype and karyotype over several passages in culture, suggests that CB-MSCs may represent a safe and readily available source for regenerative approaches in the clinical arena. Further refinements of the method are warranted to improve yield and function, but the data herein reported suggest that cells easily obtained from human CB may be a suitable source to derive insulin-producing cells for the treatment of diabetes.
Footnotes
Acknowledgments
We are very grateful to the people at the New York Blood Bank for providing the cord blood specimens that made this work possible; they have shown continuous support and dedication in helping research at the DRI, and we are deeply indebted. We wish to thank George McNamara, director of the DRI Imaging Facility for his assistance in the analysis of cells with fluorescent microscopy. We are also grateful to the staff of the DRI animal core facility for their help with the use of murine models of transplantation. This work was supported by the Diabetes Research Institute Foundation.
The following author contributions are recognized: Conception and design, all laboratory experiments for in vitro and in vivo studies, collection and/or assembly of data, data analysis and interpretation, manuscript writing (K.R.P). Data analysis and interpretation, manuscript writing (J.D.B.) Collection and/or assembly of data, data analysis, and manuscript writing for in vivo experiments (R.D.M). Data analysis and interpretation, manuscript writing (A.P). Collection and/or assembly of data for in vivo experiments (S.V.). Financial support, final approval of manuscript (C.R.). Conception and design, data analysis and interpretation, manuscript writing (L.I). The authors declare no conflict of interest.