
Abstract
This study compares mesenchymal cells isolated from excised burn wound eschar with adipose-derived stem cells (ASCs) and dermal fibroblasts in their ability to conform to the requirements for multipotent mesenchymal stem cells (MSCs). A population of multipotent stem cells in burn eschar could be an interesting resource for tissue engineering approaches to heal burn wounds. Cells from burn eschar, dermis, and adipose tissue were assessed for relevant CD marker profiles using flow cytometry and for their trilineage differentiation ability in adipogenic, osteogenic, and chondrogenic conditions. Although the different cell types did not differ significantly in their CD marker expression, the eschar-derived cells and ASCs readily differentiated into adipocytes, osteoblasts, and chondrocytes, while dermal fibroblasts only exhibited some chondrogenic potential. We conclude that the eschar-derived mesenchymal cells represent a population of multipotent stem cells. The origin of the cells from burn eschar remains unclear, but it is likely they represent a population of adult stem cells mobilized from other parts of the body in response to the burn injury. Their resemblance to ASCs could also be cause for speculation that in deep burns the subcutaneous adipose tissue might be an important stem cell source for the healing wound.
Keywords
Introduction
Due to the progression in burn wound care of recent decades the percentage of patients surviving their burn injury has greatly increased. The increased survival rate has also increased the number of people who are left with burn scars that are often cause for great functional problems due to contractures. Also their disfigurement can cause psychosocial problems (33). These developments have induced new research into wound healing with a focus on the prevention/reduction of scar formation. The most promising approach to these problems is tissue engineering, which aims at rebuilding the lost skin. This is achieved by applying differentiated skin cells or stem cells to a scaffolding material that will induce those cells to organize into a normal skin configuration. Two major problems with the tissue engineering approach are the scaffold design and the source of cells. The scaffold should create a microenvironment that will guide the cells towards tissue regeneration. Despite the enormous number of different scaffold materials developed recently, fundamental knowledge about exact composition and architecture necessary for skin tissue engineering is still scarce. Hence, clinical improvements on the conventional split skin graft treatment are limited (32).
The choice of cells for skin tissue engineering is also vital to the outcome of the wound healing process. Autologous healthy skin cells would obviously be preferable and some significant results have been achieved in supplying epidermal cells to wounds (5,35). Providing dermal cells for tissue engineering is more problematic, as conventional split skin grafts contain only a small dermal component. This obviously adds to the more well-known problems of skin grafting, namely that the creation of additional wounds as donor sites is not ideal as it is associated with pain and morbidity; furthermore, in extensive burns donor sites might be limited.
The discovery of adult stem cells in different tissues in recent years has provided researchers with an alternative source of cells for tissue engineering purposes without having to disturb the patients remaining healthy skin. It has been shown that mesenchymal stem cells (MSCs) isolated from bone marrow can be used to stimulate wound healing (9,11,36). However, the harvesting of stem cells from bone marrow is a painful procedure that yields only limited amounts of multipotent cells. Recently attention has turned more towards stem cells isolated from subcutaneous fat, which is far more abundant, easily accessible, and yields higher numbers of multipotent cells than bone marrow (39). These adipose-derived stem cells (ASCs) have been tested in a variety of in vitro and in vivo models resembling various injuries. They were shown to improve the healing process compared to control treatments (21). With regard to wound healing, experiments have mainly focused on the wound closure time in rodents, where it was shown that ASCs were able to improve the rate of wound closure (1,14,24).
Burn eschar is the nonviable tissue that remains after a burn injury. This tissue is essentially dead and surgical removal of the eschar is known to decrease the incidence of infection, reduce the inflammatory response, and improve the outcome of the wound healing process (4,12). Our laboratory has isolated mesenchymal cells from eschar tissue after debridement of a burn wound and proposed to use this cell population for tissue engineering purposes (34). Our research into these eschar-derived mesenchymal cells revealed that they were highly similar to mesenchymal cells derived from subcutaneous fat in both α-smooth muscle actin (α-SMA) expression and their ability to contract collagen scaffolds in vitro. Also fluorescence-activated cell sorting (FACS) analysis did not reveal differences between the two populations (31). The mesenchymal cells derived from subcutaneous fat, described in the latter article, were later identified as stem cells (39). We suspect that the mesenchymal cells isolated from burn eschar might actually also be MSCs that have migrated into the wound area from the subcutaneous fat or another source of MSCs after burn injury.
In this study we investigated whether eschar-derived cells fulfill all the criteria, formulated by the International Society for Cellular Therapy (ISCT) for multipotent mesenchymal stromal cells. We assessed the differentiation potential of mesenchymal stromal cells isolated from normal dermis, adipose, and burn eschar tissue and examined these cell types for an array of cluster of differentiation (CD) markers that have been postulated to be indicative of multipotent mesenchymal stromal cells (8).
Materials and Methods
Tissue Handling and Cell Culture
Mesenchymal cells derived from dermis and subcutaneous fat were isolated from tissue obtained from healthy donors during abdominal dermolipectomy. A split skin biopsy 0.3 mm in thickness was removed using a dermatome (Aesculap AG & Co. KG, Tuttlingen, Germany). Subcutaneous fat samples were cut from the material using sterile scissors. Eschar material was obtained from patients undergoing burn treatment in the burn center of the Red Cross Hospital Beverwijk, Netherlands. Eschar tissue was removed between 11 and 26 days post-burn injury. All tissue was obtained in accordance with the guidelines of the Red Cross Hospital, Beverwijk and according to the code of proper use of human tissue formulated by the federation of Dutch medical scientific societies (FMWV/FDMSS).
Cells were isolated from the different types of tissue as described previously (31). Dermis and epidermis were separated enzymatically by incubating in a saline solution with 0.25% dispase II (Boehringer Mannheim, Germany) at 37°C for 1 h. Dermal, fat, and eschar tissues were washed, weighed, and minced with sterile scissors. Materials were then incubated for 1.5—3 h in 2 ml of saline containing 0.25% dispase II/collagenase A (Boehringer, Mannheim, Germany) for every gram of tissue at 37°C under continuous agitation. The resulting suspension was passed through a filter chamber (Beldico, Duiven, Netherlands) and washed with 1% fetal calf serum (FCS; Hyclone, Logan, UT) in phosphate-buffered saline (PBS; Gibco, Pastley, UK). Cells were pelleted, suspended in fibroblast culture medium (FBM) [DMEM with 10% FCS, 1 mM <sc>l</sc>-glutamine, 100 μg/ml streptomycin, and 100 IU/ml penicillin (Gibco, Pastley UK)] passed through a 0.7-μm cell strainer (Fischer, Landsmeer, Netherlands), pelleted again, and seeded at approximately 20,000—30,000 cells/cm2. One day after isolation the cells were washed with PBS and fresh culture medium was added. Cell passage was performed using trypsin (Gibco, Pastley, UK) just prior to reaching full confluence.
In total 13 cell populations isolated from dermis (age: 26—51 years, mean: 38 years), 9 from adipose tissue (age: 33—51 years, mean: 42 years), and 12 from eschar tissue [age: 8 months—79 years, mean: 38 years, total body surface area (TBSA): 0.5—19%, mean: 4%, eschar excision post burn: 11—26 days, mean: 14 days] were used in this study.
For flow cytometry, the following number of populations were analyzed: dermis (n = 10), adipose (n = 8), eschar (n = 7). For general trilineage differentiation: dermis (n = 3), adipose (n = 4), eschar (n = 4). For micromass culture: dermis (n = 3), adipose (n = 3), eschar (n = 4).
Flow Cytometry
After three passages cells were labeled with fluorescently labeled specific antibodies (Table 1) for flow cytometry. Cells were trypsinized and washed in facs buffer (0.1% bovine serum albumin; Sigma-Aldrich, St. Louis, MO) and 0.05% sodium-azide in PBS). Samples were then incubated with the antibodies in Table 1 for 30 min at 2—8°C. Afterwards cells were washed with FACS buffer twice and stored in FACS buffer until being measured on a Facscalibur flow cytometer (BD Biosciences, San Jose, CA), within 24 h after labeling. Cells were kept on ice or at 2—8°C during the whole procedure.
Antibodies Used in Flow Cytometry

CD, cluster of differentiation; FITC, fluorescein isothiocyanate; LL-FITC, Lightining-Link FITC conjugation kit (Innova Biosciences); PE, phycoerythrin, HLA, human leukocyte antigen.
CD73 antibody was conjugated with fluorescein isothiocyanate (FITC) using the lightning-link conjugation kit (Innova biosciences, Cambridge, UK) in accordance with the manufacturer's specifications. Data analysis was performed with FCS express 3 software (De Novo software, Los Angeles, CA). The percentage of positive cells in each sample was calculated by Overton histogram subtraction (22).
Differentiation Assay
After four passages cells were used to assess their differentiation potential in the adipogenic, chondrogenic, and osteogenic lineages. Cells were trypsinized at approximately 90% confluence and seeded into Lab-Tek chamberslides (Fischer, Landsmeer, Netherlands) (5,000 cells/cm2). Cells were cultured in standard fibroblast culture medium until almost reaching full confluence; the culture medium was then replaced by the appropriate Miltenyi NH differentiation media (Miltenyi biotec, Bergisch Gladbach, Germany). Cells were maintained in this medium for 2 weeks (v. Kossa samples for up to 4 weeks); medium was changed twice a week.
Micromass Culture
Chondrogenic differentiation was investigated in micromass culture. Cells were placed in centrifuge tubes at a density of 0.3 × 106 cells/tube and centrifuged at 350 x g for 5 min. Cells were placed in culture as a pellet in chondrogenic differentiation medium (Miltenyi biotec, Bergisch Gladbach, Germany) and FBM. Medium was changed twice a week, and the resulting cell mass was fixed in 4% formalin for 24 h after 3 weeks of culture, dehydrated, and embedded in paraffin for histological examination.
Staining for Differentiated Cells
Staining for differentiated cells was adopted from earlier protocols with some minor adjustments (30,39).
Osteogenic Differentiation: Alkaline Phosphatase
Slides were incubated with New Fuchsin (Dako, Glostrup, Denmark) for 30 min at 37°C. Staining was stopped by adding FBM and cells were fixed with 4% formalin at 4°C for 30 min. Slides were rinsed with PBS and counterstained with hematoxylin.
Osteogenic Differentiation: Von Kossa
Cells were fixed for 1 h in 4% formalin at 4°C and then incubated in a 1% silver nitrate solution under ultraviolet light for 1 h. Afterwards unreacted silver was removed by incubating in 5% sodium thiosulfate for 5 min. Slides were counterstained with eosin.
Chondrogenic Differentiation: Alcian Blue (pH 1.0)
Cells were fixed for 15 min in 4% formalin at room temperature and incubated for 30 min with 1% Alcian blue (8 GX Sigma-Aldrich, St. Louis, MO) in 0.1 M HCl (pH 1.0). Slides were counterstained with nuclear fast red (Dako, Glostrup, Denmark).
Adipogenic Differentiation: Oil Red O
Cells were fixed for 1 h in 4% formalin with 1% calcium chloride. Slides were then washed in 70% ethanol and incubated for 30 min in 2% oil red O (Sigma-Aldrich) in 60% 2-propanol solution at room temperature. Slides were counterstained with hematoxylin.
Chondrogenic Differentiation: Collagen 2a1 RT-PCR. RNA Preparation
Cultures were prepared from dermis (n = 3), adipose (n = 3), and eschar cells (n = 3) cultured in chondrogenic differentiation medium [DMEM, 1% FCS, 6.25 μg/ml insulin, 10 ng/ml transforming growth factor-β1 (TGF-β1), 50 nM ascorbate-2-phosphate, 1% antibiotics, as described in Zuk et al. (39)]. RNA was isolated using TRIzol reagent (Invitrogen Life Technologies, Breda, The Netherlands) according to the manufacturer's instructions.
Real-Time RT-PCR Assays
RNA (500 ng) was reverse transcribed using the QIAGEN QuantiTect® reverse transcription kit according to the manufacturer's instructions. The resulting cDNA was diluted to 100-μl and 5-μl samples, which were used for amplification in PCR reaction.
Real-time RT-PCR was performed using the iQ SYBRgreen Supermix (Bio-Rad, Veenendaal, The Netherlands). cDNA was amplified using specific collagen 2a1 primers (forward: GGAGCAGCAAGAGCAAG GAGAAG; reverse: TGGACAGCAGGCGTAGGAAGG) at a concentration of 0.40 μM, in an iCycler iQ thermal cycler (Bio-Rad). After activating the DNA polymerase by incubation for 2 min at 95°C, 40 cycles of amplification (95°C for 30 s and 55°C for 60 s) were performed. Fluorescence was monitored during every thermal cycle at the 55°C annealing step. After PCR, the baseline subtraction method was used to determine the threshold cycle.
Data are expressed as ratios between target mRNA and housekeeping genes β2-microglobulin (forward: GGCATTCCTGAAGCTGAC; reverse: ATGTCGGAT GGATGAAACC) and YWHAZ (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide; primers forward: AGCAGAGAGCA AAGTCTTC; reverse: GCTTCTTGGTATGCTTGTTG).
Statistical Analysis
Data were analyzed by SPSS software version 16.0. Expression is presented relative to the control condition, which is set at 1. Data are displayed in box plots. The boxes represent the middle 50% of the values (25th—75th percentile), the line in the box represents the median value, and the bars the highest and lowest value. Statistical analysis was performed nonparametrically using the Mann-Whitney test (p < 0.05).
Results
Flow Cytometry
Flow cytometry shows that all three cell populations used in this study meet the mesenchymal stromal cell definitions. They are positive for CD90, CD105, and CD73 and negative for CD31, CD45, CD14, CD79a, and human leukocyte antigen (HLA)-DR. However, a small percentage of cells was found to express CD34. This resembled a small shift of the population and not a clearly positive subpopulation of cells. Differences in CD34 expression did not seem to be correlated with donor age, time between burn, and eschar removal or total body surface area (TBSA).
Table 2 presents the mean positive percentages calculated by Overton histogram subtraction for each CD marker.
Flow Cytometry Results Expressed as the Mean Percentage of Positive Cells Followed by the SD

In the criteria formulated by the International Society for Cellular Therapy (ISCT) cells should express CD90, CD105, and CD73 and not express CD31, CD34, CD45, CD14, CD79a, or HLA-DR. CD34 expression was highly varied, but not unexpected for cultured mesenchymal cells. CD271 expression is reported to be associated with increased proliferation and differentiation potential; however, we did not find any noticeable expression in the cell populations we tested. ASC, adipose-derived stem cells.
Figure 1A—D shows representative histogram profiles of ASCs and eschar-derived cells as well as examples of variation in CD34 expression between samples of the same cell type.

Flow cytometry histograms for relevant cluster of differentiation (CD) markers. Control histograms in black and sample histograms in red. (A) Representative flow cytometry histograms for adipose-derived stem cells (ASCs). (B) Variation of CD34 expression between different ASCs samples. (C) Representative flow cytometry histograms for eschar-derived cells. (D) Variation of CD34 expression between different eschar samples.
Differentiation
Differentiation was assessed through four different stains. Alcian blue (pH 1.0) stains highly sulfated mucosubstances, which are one of the main components of cartilage and are used to demonstrate the presence of chondrocytes. Alkaline phosphatase is usually found in liver or bone tissue and used along von Kossa staining to determine osteogenic differentiation. von Kossa staining visualizes calcium or calcium salts, indicative of bone formation. Oil red O staining is used to demonstrate the presence of lipids, indicative of adipocyte differentiation. Mesenchymal cells isolated from adipose tissue and eschar tissue differentiated successfully towards osteogenic, chondrogenic, and adipogenic lineages. However, cells from normal dermis were unable to differentiate towards osteogenic and adipogenic lineages. However, micromass culture of these cells did produce some positive Alcian blue staining, indicative of chondrogenic differentiation capability.
Figure 2 shows representative images of the different stains performed to establish differentiation.
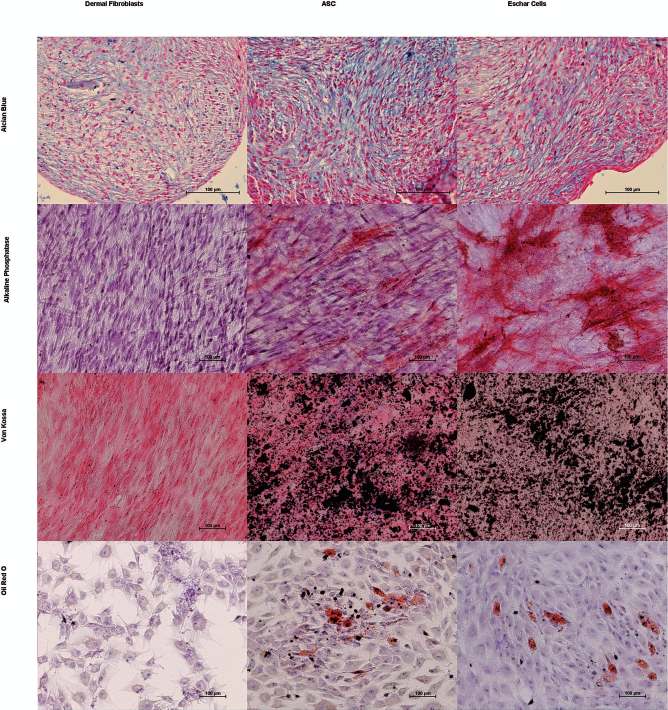
Representative images from differentiation experiments. Columns from left to right show results for dermal fibroblasts, ASCs, and eschar-derived cells. Rows from top to bottom show Alcian blue staining (sulphated mucosubstances in blue), alkaline phosphatase (enzymatic activity in red), von Kossa staining (calcium deposits in black), and Oil red O staining (lipid deposits in red). ASCs and eschar-derived cells clearly differentiate into all three lineages, while dermal fibroblasts only appear to show some chondrogenic differentiation ability. Scale bars: 100 μm.
Collagen 2a1 RT-PCR
PCR results are displayed in Figure 3. All three cell types used in this study expressed significantly more collagen 2 RNA when cultured in chondrogenic differentiation medium compared to FBM control samples (Mann-Whitney p < 0.05).
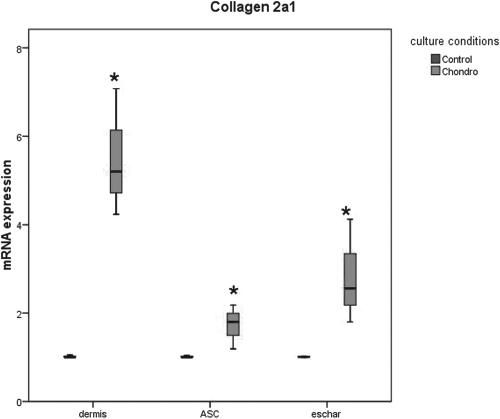
mRNA expression levels of collagen 2a1 in cells after chondrogenic differentiation. mRNA values are expressed relative to housekeeping genes β2-microglobulin (β2M) and YWHAZ and presented as box plots. *All cell types used had significantly higher collagen 2a1 expression when cultured on chondrogenic differentiation medium instead of fibroblast culture medium (FBM; Mann-Whitney p < 0.05).
Discussion
In this article we describe that the mesenchymal cells isolated from burn eschar meet the criteria of multipotent mesenchymal stromal cells as defined by the ISCT (8). Cells isolated from dermal tissue showed limited differentiation capacity, even though they did match the required CD marker profile. ASCs did not perform different from the eschar-derived cells in any of the experiments performed in this study. Therefore, we refer to the eschar-derived cells as burn eschar-derived mesenchymal stem cells (BESCs).
Quirici et al. linked CD271 expression on ASCs to increased proliferation and differentiation potential (23). We included CD271 in our analysis to see if we could identify a CD271-positive population in our cell populations. In all three of our cell populations CD271 expression was very low (1—2%) but nevertheless the differentiation potential of the ASCs and BESCs could be established. CD34 expression varied widely between cells isolated from different types of tissue. However, variation was also observed between different samples of cells isolated from the same type of tissue. Changes in CD34 expression were always observed as shifts in the mean fluorescence of the entire cell population and not as distinct positive and negative populations. According to the ISCT criteria MSCs should not express CD34 as this is a hematopoietic cell marker. However, the gradual loss of CD34 expression by ASCs in culture is an often observed phenomenon (2,20,27,37). A recent study has established that although CD34 expression is correlated with differences in proliferation and differentiation potential, it is not a marker that is essential to the identification of ASCs. CD34 expression of ASCs is even reported to be reversible depending upon culture conditions (26).
Although dermal fibroblasts have a similar CD marker expression pattern as multipotent mesenchymal stromal cells, their differentiation potential is limited to the chondrogenic lineage and this cell population probably contains only very few stem cells. Other groups already demonstrated that although FACS analysis performed with the described CD markers show a more or less homogenous cell population, only a small percentage of these cells are multipotent stem cells (13). These results show that flow cytometry data need to be accompanied by trilineage differentiation to determine whether a cell population contains MSCs. Further research will be needed to identify, to date undiscovered, discriminating markers or a more refined set of known CD markers that will successfully identify MSCs without the need for differentiation experiments.
MSCs exhibit a contractile phenotype. In studies culturing bone marrow-derived mesenchymal stem cells (BMSCs) on dermal scaffolds (25) as well as in our own experiences with ASCs and BESCs in such applications (31), the cells contract the scaffold and are found to express αSMA. In human wound healing, contraction and αSMA expression are associated with myofibroblast differentiation. These cells are generally associated with poor wound healing, as they produce excessive and abnormal extracellular matrix (ECM) and are held responsible for the contractures often seen in hypertrophic scars (6). Even though αSMA expression is not a definitive marker of myofibroblasts, contraction data suggest that mesenchymal stromal cells are able to differentiate into myofibroblasts. On the other hand, αSMA expression could be a property of MSCs unrelated to myofibroblast differentiation. Strikingly, all multipotent mesenchymal cell populations that seem to have a high differentiation potential contain high percentages of αSMA-positive cells. Suga et al. also reported that CD34-negative ASCs displayed greater differentiation potential than CD34-positive ASCs, as well as increased αSMA RNA expression, suggesting a link between increased αSMA expression and differentiation potential (26).
The origin of the BESCs is open to speculation. The eschar tissues used in this study were excised between 11 and 26 days after the burn injury. This is done to allow partial thickness burns to heal without the need for skin grafts, according to the policy of many European burn centers. Delayed surgical excision combined with specific topical treatment is a regular treatment option in the Netherlands. During this period the necrotic tissue or the severely damaged tissue could attract (stem) cells from the surrounding tissues, through chemokine secretion by inflammatory cells, in an attempt to heal the defect. The BESCs could possibly be ASCs or another adult population of MSCs migrating from their respective tissues to the site of the burn injury. Several studies have shown that stem cells from bone marrow seem to be able to home to the wound area and integrate in the regenerated skin, both in the dermis and epidermis (10,24,36). Elevated levels of circulating MSCs have also been detected in the blood of burn victims (17). However, a recent study has demonstrated that bone marrow-derived cells probably do not contribute to the fibroblast and myofibroblast population of a healing wound in mice (3). But the subcutaneous adipose tissue could still be considered a potential source of multipotent mesenchymal cells, which could play a role in the healing of deep skin wounds.
Evidence for the involvement of the subcutaneous fat in wound healing is provided by the presence of fat domes in anatomical locations prone to hypertrophic scarring (18). The role BESCs play in wound healing is unclear, as is the timing of the influx into the wound area. It is very likely that these cells contribute to the closure of the defect, as they possess the myofibroblast phenotype that plays an important role in ECM production and wound contraction. We have tried to isolate BESCs from some early excisions that are performed in our local burn center, but have been unable to detect them in reasonable numbers in these eschar samples. Although we have not determined at what time postburn BESCs generally appear, this suggests that it might take some time for them to migrate into the burned tissue. It is very well possible that the influx of BESCs continues after excision of the eschar and that these cells will influence the healing process also after the application of the skin graft. Regardless of the timing of influx of these cells, we think this finding represents an interesting option for novel wound treatment, because it offers the possibility to isolate the BESCs from a late excision eschar and use them in tissue engineering applications for burn wounds.
MSCs can only be expected to be beneficial to tissue regeneration if they differentiate into the locally required phenotype in the wound environment. If the correct signals from normal skin are absent the application of MSCs to skin wounds may actually be detrimental to the quality of the healing process. If MSCs can indeed differentiate into myofibroblasts, their application to full thickness wounds in humans might therefore actually lead to more scar formation. Unfortunately, most in vivo stem cell applications in wound healing have been performed in rodents, where fast wound closure due to contraction is considered a sign of improved wound healing (1,11,14,24,36). However, in humans and experimental animals with skin that resembles human skin more closely, like pigs (19,28), increased wound contraction is generally considered a negative outcome of wound healing. A study using stromal cells from adipose tissue in dermal substitutes in pig wounds indicates this may indeed be the case. This application of ASCs “avant la letter” demonstrated that seeding the substitutes with dermal fibroblasts significantly reduced wound contraction, while the addition of adipose-derived cells did not reduce contraction compared to unseeded controls (7). Furthermore, this study demonstrated that the number of αSMA-positive cells and wound contraction could be significantly reduced by removing vascular fragments from the adipose tissue fraction prior to cell isolation and seeding of the dermal substitute. Recent studies have provided evidence that the stem cell niche in adipose tissue is probably the adipose vasculature (16,38). This suggests that by removing vascular fragments the number of mesenchymal stem cells seeded into scaffolds was greatly reduced. Klinger et al. demonstrated that injecting ASCs under matured scar tissue could improve the facial mimicry, skin texture, softness, thickness, and elasticity of the scar (15). This demonstrates that when skin homeostasis is somewhat restored (albeit in the form of a scar) the behavior of ASCs may actually be very different compared to their actions in a wound environment. The relevance of stem cells in lipofilling has recently been reviewed (29).
More research is required to elucidate both the origins of the stem cells we have identified in burn eschar, their possible link with myofibroblasts, and how their function is influenced by the wound environment. Elucidating which signals are required to differentiate MSCs into the various skin cells, and away from a contractile phenotype, might allow medical science to design effective strategies for their future use in tissue regeneration.
Footnotes
Acknowledgments
We would like to thank the following people for their contributions to this study: Tom O'Tool, for his assistance with our flow cytometry work and Antoon van den Bogaerdt and Michelle Verkerk for their preliminary work that led to this article. The authors declare no conflicts of interest.