
Abstract
Cotransplantation of neural progenitors (NPs) with Schwann cells (SCs) might be a way to overcome low rate of neuronal differentiation of NPs following transplantation in spinal cord injury (SCI) and the improvement of locomotor recovery. In this study, we initially generated NPs from human embryonic stem cells (hESCs) and investigated their potential for neuronal differentiation and functional recovery when cocultured with SCs in vitro and cotransplanted in a rat acute model of contused SCI. Cocultivation results revealed that the presence of SCs provided a consistent status for hESC-NPs and recharged their neural differentiation toward a predominantly neuronal fate. Following transplantation, a significant functional recovery was observed in all engrafted groups (NPs, SCs, NPs + SCs) relative to the vehicle and control groups. We also observed that animals receiving cotransplants established a better state as assessed with the BBB functional test. Immunohistofluorescence evaluation 5 weeks after transplantation showed invigorated neuronal differentiation and limited proliferation in the cotransplanted group when compared to the individual hESC-NP-grafted group. These findings have demonstrated that the cotransplantation of SCs with hESC-NPs could offer a synergistic effect, promoting neuronal differentiation and functional recovery.
Keywords
Introduction
The limited endogenous capacity of the spinal cord for repair and regeneration has directed the focus of preclinical trauma research toward ways for prevention of cell death, cell transplantation, or support of degenerating neurons (8,80). A variety of cell replacement strategies have been examined for the potential to repair and regenerate the spinal cord in animal models. The most commonly used cell types include, but are not limited to, olfactory ensheathing cells (OECs) (43,72,75), bone marrow and cord blood stromal cells (16,63,66), Schwann cells (SCs) (7,9,21), and genetically modified fibroblasts (58,81). A systematic review of cellular transplantation therapies for spinal cord injuries (SCI) in different animals from rodents to primates has been performed by Tetzlaff and colleagues (79). Accordingly, neural stem cell or progenitor (NSC/NP) transplantation is one promising strategy for promoting functional recovery from SCI by increasing survival and growth of the host tissue, and/or replacement of lost cells primarily due to their self-renewal and given their ability to generate all types of neural cells (27,49). These cells are derived from in vivo fetal/adult tissues (65) or pluripotent stem cells such as human embryonic stem cells (hESCs) in vitro (20,24,34,37,45,76). hESCs provide a useful source of cells for basic developmental studies and cell-based therapies because of their unlimited self-renewal and pluripotency capacities (31). So far, many promising studies have shown the therapeutic potential of hESC-derived NPs (hESC-NPs) to ameliorate neurological diseases in animal models such as Parkinson's disease (5,73) and retinal macular degeneration (30,52). Moreover, grafting of ESCs into animal models with SCI is shown to improve motor function (15,23,42,54). However, there are two major problems with the use of NP implants for the treatment of SCI: a poor survival rate and the low rate of differentiation of NPs into neuronal cells (12,88).
One way to overcome these problems is the cotransplantation of cells, which has been tested to promote recovery in animal models of SCI (1,22,39,47,56,74, 84,91). To date, cotransplantation of NSCs with basic fibroblast growth factor (bFGF)-expressing amniotic epithelial cells (56) and olfactory ensheathing cells (OECs) (84) has been reported. Additionally, a few studies using in vivo-derived NPs and intact SCs or genetically modified SCs have induced functional recovery and achieved good neuronal differentiation of grafted cells in animal models of SCI (47,62,90,91), thus providing mounting evidence for the idea that SCs may contribute to survival and neuronal differentiation of NPs in vivo.
In the present study, initially we generated NPs from hESCs. hESC-NPs provide unlimited availability for NPs and overcome teratoma formation, one of significant hurdles in hESC technology. These NPs were then cocultured with SCs isolated from rat sciatic nerve with the intent to evaluate the effect of SCs on neurogenesis in vitro. It was determined that SCs increased neuronal differentiation of hESC-NPs in vitro. Then, to examine the potential effect of SCs on promoting the in vivo neuronal differentiation fate and functional recovery of hESC-NPs, we transplanted cultured SCs along with NPs into contused injured spinal cords of adult rats.
Materials and Methods
Culture of hESCs and Generation of NPs
The hESC line Royan H6 (passage 45) (2) was used in these experiments. Cells were expanded and passaged under feeder-free culture conditions in hESC medium that contained 100 ng/ml bFGF as previously described (57). The medium was changed every other day, until day 7. To generate NPs from feeder-free hESCs, we employed a protocol that used a combination of retinoic acid (RA; 2 μM, Sigma-Aldrich), bFGF (100 ng/ml, Royan institute), and noggin (250 ng/ml, R&D) in DMEM-F12 medium as previously described (59).
The hESC-NPs were passaged using 0.008% trypsin (Invitrogen)/2 mM ethylenediaminetetraacetic acid (EDTA; Merck) (17) on laminin (1 mg/ml) and poly-L-ornithine (15 mg/ml, Sigma-Aldrich)-coated tissue culture dishes in the same medium, which contained 20 ng/ml epidermal growth factor (EGF) and 20 ng/ml bFGF as NP expansion medium. Media were changed every other day for 5—7 days. All experiments were performed by hESC-NPs at passage 11—15. Spontaneous differentiation was performed in differentiation medium in the absence of growth factors, including: neurobasal medium (Invitrogen)/DMEM-F12 (1:1), B27 (1%), knockout serum replacement (KOSR, 5%), and N2 supplement (1%) for 35 days. Half of the medium was renewed every 5 days.
Immunocytofluorescence Staining
The cells were fixed in 4% paraformaldehyde or ethanol for 20 min at room temperature and −20°C, respectively, permeabilized with 0.2% Triton X-100 for 5 min, and blocked in 10% host (goat/rabbit) serum in PBS for 1 h. Cells were incubated with primary antibodies (Table 1) for 1 h at 37°C, washed, and incubated with secondary antibodies (Table 1) as appropriate for 1 h at 37°C. The nuclei were counterstained with propidium iodide (PI; Sigma, P4170) or 4,6-diamidino-2-phenylindole (DAPI; 1 μg/ml, Sigma-Aldrich, D-8417) and/or 1 μg/ml PI (Sigma-Aldrich, P4864) for 3 min at room temperature. Omission of the primary antibody in the sample was used as a control for all markers. Labeled cells were examined with a fluorescent microscope (Olympus, BX51, Japan) and images were acquired with an Olympus D70 camera.
Antibodies Used in This Study

Sox1, sex determining region Y-box 1; Pax6, paired box gene 6 (aniridia, keratitis); P75LNGFr, p75 low-affinity nerve growth factor receptor; FITC, fluorescein isothiocyanate; TRITC, tetramethyl rhodamine isothiocyanate.
Flow Cytometry Analysis
All staining reactions were performed in staining buffer consisting of PBS supplemented with 1% fetal bovine serum (FBS) and 0.085% EDTA (Sigma, E6758). After determination of the cellular viability by trypan blue exclusion, cells were washed twice in staining buffer and fixed in 4% paraformaldehyde or ethanol. Triton X-100 (0.2%) was used for permeabiliztion. Nonspecific antibody binding was blocked with a combination of 10% heat-inactivated goat serum (SAFC Biosciences, 12036-500M) in staining buffer and 1.5—2.0 × 105 cells were used per sample. Cells were incubated with the appropriate primary antibodies or appropriate isotype-matched controls. Primary antibodies used were: anti-nestin, anti-Sox1 (sex determining region Y-box containing gene 1), and anti-p75 low-affinity nerve growth factor receptor (P75LNGFr) (Table 1). The cells were washed two times in staining buffer and incubated for 30 min at 4°C with the appropriate secondary antibody (Table 1). After washing, flow cytometric analysis was performed with a BD-FACS Calibur Flow Cytometer. The experiments were replicated three times and the acquired data were analyzed with WinMDI (2.9) software.
Karyotype Analysis
Karyotype analysis was done as described elsewhere (57). Briefly, the cells were treated with thymidin (0.66 μM, Sigma) for 16 h and then with colcemid (Gibco, 0.15 μg/ml, 30 min). Subsequently, the cells were exposed to 0.075 M KCl at 37°C for 16 min and fixed three times with ice-cold 3:1 methanol/glacial acetic acid and dropped onto precleaned chilled slides. Chromosomes were visualized using standard G-band staining. At least 20 metaphase spreads were screened and 10 of them were evaluated for chromosomal rearrangements.
RNA Isolation and Real-Time RT-PCR for Gene Expression Analysis
The expression patterns of specific genes were studied and compared with undifferentiated hESCs. From each sample, total RNA was extracted using RNX reagent (Cinnagen, RN7713C). RNA (5 μg) was treated with DnaseI (Fermentas). cDNA was synthesized from 1 μg of RNA using the RevertAid™ H Minus First Strand cDNA Synthesis Kit (Fermentas) with a random hexamer primer. The resulting cDNA were subjected to real-time RT-PCR. Specific human primers Oct4 [octamer binding protein 4; POU domain, class 5, transcription factor 1(Pou5F1)], Nanog (Nanog homeobox), Otx2 (orthodenticle homeobox 2), Hoxa2 (homeobox a2), Hoxa5, Hoxb4, and Hoxc5 were designed with Perl-primer V.1.1.14 software. Real-time RT-PCR was carried out using a 96-well optical reaction plate and a 7500 real-time PCR system (Applied Biosystems). For each PCR run, 40 ng cDNA products were mixed with 1x SYBR Green PCR Master Mix and PCR primers in a total volume of 20 μl. Relative gene expression was analyzed using the comparative Ct method, 2−ΔΔCt (50). Target genes were normalized by the reference gene, β-actin (31), and calibrated for each sample against control cells (undifferentiated hESCs). Each experiment included three replicates and each replicate consisted of two identical samples. Thermal conditions for all genes were the same and the annealing temperature was 60°C.
Animals
Subjects of this study were Wistar rats (Pasteur Institute, Tehran, Iran) weighing about 250—300 g. All animal care, surgical processes, and postoperative euthanasia were undertaken in strict accordance with the approval of the Institutional Review Board and Institutional Ethical Committee. All efforts were made to minimize the number of animals used. Animals were housed two per cage and provided free access to food and water throughout the study. All surgical procedures were performed under anesthesia produced by a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg) (61).
Isolation, Culturing, and Labeling of SCs
SCs were isolated from adult rats' sciatic nerve bilateral biopsies according to established protocols (60). In brief, 2 weeks after in vitro predegeneration, sciatic explants were dissociated and plated onto 2 μg/ml laminin-coated dishes in DMEM medium. Finally, the medium was changed with melanocyte growth medium (MGM, PromoCell, C-24300) to which 2 μM forskolin (Calbiochem, 344273), 10 ng/ml FGF2 (Sigma, F0291), and 5 μg/ml bovine pituitary extract (BPE-26, Promo-cell, C-30021) were added in order to prevent fibroblast proliferation. Ethylene glycol tetraacetic acid (EGTA) purification was implemented after 5—6 days and the supernatant was collected and transferred to new dishes. The purity of SCs for grafting was greater than 99%. Later passages (passages 3—4) were used in the coculture system.
PKH fluorescent dye was used to label SCs before transplantation. According to its protocol, adherent SCs were suspended in a 2x suspension of the respective cells and a 2x dye solution, both in the PKH diluent supplied with the kit (PKH26-GL, Sigma), mixed, and incubated for 3 min. The labeling reaction was stopped by addition of medium plus 10% serum. Labeled SCs were washed three times to remove unbound dye. The stable partitioning of the fluorescent dye into the membrane permits long-term monitoring while leaving the important functional surface proteins unaltered. The labeled SCs (passages 3—4) were transplanted into contusion models of SCI.
Coculture of hESC-NPs and SCs
For coculture, approximately 3 × 104 hESC-NPs were added onto the SCs. In the control group, hESC-NPs were seeded onto poly-l-ornithine (15 μg/ml, Sigma, P4957) and laminin (15 μg/ml, Sigma, L2020)-coated coverslips in expansion medium for 24 h to improve attachment and allow for a consistent cell status. Subsequently, the medium was switched to differentiation medium consisting of a mixture of DMEM/F12 neurobasal medium (1:1) with 5% KOSR, 1% N2, 1% B27, 2 mM l-glutamine, 1% nonessential amino acids, 100 U&/ml penicillin, and 100 μg/ml streptomycin. Half of the medium was changed every 3—4 days and continued for 3 weeks. Next, fixation and immunostaining were implemented for the TUJ1 (β-III-tubulin antibody), MAP2 (microtubule associated protein 2), and GFAP (glial fibrillary acidic protein) markers.
Preparation of Cells for Transplantation
For identification after transplantation, we utilized human nuclear antigen (HNu). HNu is a selective nuclear marker of cells originating from humans. Therefore, it was not necessary to label hESC-NPs. SCs were prelabeled with PKH-26 (PKH26-GL, Sigma), which allowed for the identification of grafted cells in vivo. Immediately before transplantation the respective cells were harvested and washed three times in PBS. A sample of the resulting single cell suspensions was stained with trypan blue dye exclusion (Sigma) and counted with a Neubauer hemocytometer. hESC-NP and SC suspensions were mixed and diluted in PBS to yield the appropriate cell densities for the various grafting paradigms.
Surgical Procedure, Experimental Grouping, and Transplantation
SCI Model. Prior to any surgical manipulation, all animals were trained for the Basso Beattie Bresnahan (BBB) test (see below) for 4 days with two repeated trials per day. Weight-drop contusion injury was carried out according to Gruner (29) and Widenfalk et al. (86). Briefly, following an incision through the skin, subcutis, and muscle, a T9—10 laminectomy was performed. Next, the vertebral column was stabilized by clamps attached to the spinal processes, cranial and caudal to the laminectomy. For the contusion injury a 10-g rod was raised to a height of 25 mm above the dorsal surface of the spinal cord. It was then released and accelerated by the force of gravity until it hit the exposed spinal cord. Afterwards, the surgical wounds were sutured, and the animals were given postoperative analgesia and Ringer's solution (5 ml, SC) to prevent dehydration. Animals were allowed to recover and were housed in standard rat cages at a temperature of 27°C. Their bladders were manually expressed three times daily until return of reflexive bladder control.
Experimental Grouping and Transplantation
Forty-eight rats with normal locomotor performance were selected. The sham group (n = 8) had only laminectomy whereas rats in the other group were subjected to SCI. Rats with mean BBB scores of 1, indicating slight movement of one or two joints, were randomly assigned into five groups 1 week after SCI. The injury control group (n = 8) received no manipulations. The vehicle transplanted group (n = 8) received 5 μl PBS in the epicenter of the contusion. hESC-NP transplantation (n = 8) and SC transplantation groups (n = 8) each received 0.5 × 106 hESC-NP and SC suspensions in 5 μl PBS, respectively. The hESC-NP/SC cotransplantation group (n = 8) received 0.5 × 106 of each of the aforementioned cells or, in total, 1 million cells in 5 μl PBS. The viability of cells before transplantation was more that 97% by trypan blue exclusion dye assay.
For cell transplantation into the spinal cord lesions a 25 μl Hamilton syringe connected to a 27-gauge needle through a glass capillary (inner diameter ~50 μm) was used. A Kopf microstereotaxic injection system (David Kopf Instruments) infused the suspension at a rate of 0.5 μl/min. After grafting, the syringe was raised 1 mm and left in place for 5 min to minimize cell diffusion up the needle track. Animals were given gentamycin (15 mg/kg, SC) and Ringer's solution (5 ml, SC), and kept on a heating pad until they woke. Animals were then housed in standard rat cages at a temperature of 27°C. Starting 2 days before transplantation, all groups of rats received daily injections of cyclosporine A (10 mg/kg, SC, Sandimmune, Novartis Pharmaceuticals, East Hanover, NJ, USA) to prevent rejection.
Behavioral Assessment
To compare the functional outcome of various treatments over time, gait abnormalities in rats were blindly evaluated by two observers using the BBB Locomotor Rating Scale (3). Rats were placed in an open field (75 × 125 cm) and observed for 4 min. Hind limb function was scored from 0 to 21 (flaccid paralysis to normal gait). Performance in behavioral task was performed 3 days before injury, prior to transplantation, and continuously, once weekly, for a further 5 weeks after transplantation.
Sacrifice and Cryosection
After the behavioral analysis, all rats were given a single dosage of anesthesia, a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg), and then transcardial perfusion of anesthesia, with 0.1 M cold PBS followed by 400 ml of ice-cold 4% paraformaldehyde in PBS (pH 7.4). Rat spinal cords were dissected and postfixed at 4°C, then cryoprotected with 30% phosphate-buffered sucrose at 4°C for 96 h. Samples were successively cryosectioned in a longitudinal manner and frozen for further processing. Slice thickness was 10 μm and they were sampled at every five section intervals.
Immunohistofluorescence Staining and Quantification
For dual immunohistofluorescence staining, sections were rehydrated in 0.1 M PBS and permeabilized with 0.1% Triton X-100 (except for membranous markers) in PBS. After blocking nonspecific sites with 10% normal serum and 1 mg/ml bovine serum albumin (BSA), sections were incubated for 2 h with primary antibodies which addressed human (graft) versus rat (host) cell identity, mitotic activity, and neuron, astrocyte, and oligodendrocyte phenotype specifications. Antibodies implemented for immunohistofluorescence are presented in Table 1. For survival studies and tracing of cell fates, we utilized HNu raised in rabbits whereas the remaining primary antibodies were raised in mice. Normal IgG from the species of origin of the primary antibodies served as negative controls. After washing with PBS, cells were incubated with the appropriate fluorescein isothiocyanate (FITC) and tetramethyl rhodamine isothiocyanate (TRITC) conjugated secondary antibodies for 1 h at 37°C. Sections were washed three times with PBS and counterstained with DAPI. The coverslips were mounted onto glass slides with Entellan (Merck Germany). For quantitative determination of the differentiation pattern of transplanted cells, a fluorescence microscope (Olympus BX51, Japan) equipped with an Olympus DP70 camera was used to evaluate the number of HNu/DAPI-positive cells. For each cell marker, we used three to five tissue sections per rat (at 50 μm apart) from four rats. Then we counted the number of HNu/DAPI-positive cells that were double-labeled cells with the cell marker in 10 random fields per section. On average, 100—200 cells were counted per section.
Statistical Analysis
All data are expressed as mean ± SEM. A value of p < 0.05 was considered statistically significant. BBB scores were compared using one-way ANOVA analysis followed by Tukey's post hoc test for multiple comparisons. The Student's t-test was performed to determine statistical differences between immunostaining in hESC-NP and hESC-NP/SC cocultures and transplantation. All statistical analyses were performed on standard statistics computer software (SPSS 11.5 for Windows).
Results
Generation and Growth of hESC-NPs
Feeder-free cultured hESCs were induced to produce neural rosettes, a distinct cluster of columnar cells, and then manually harvested under phase contrast microscopy. Next, hESCs were dissociated into single cells and plated in fresh defined medium in the presence of bFGF/EGF on a laminin/poly-l-ornithine substrate. Cultures were subsequently passaged at a high cell density (1:2 ratio) every 5—7 days. These conditions generated a population with homogeneous morphology that formed multiple foci of rosette-like structures throughout the culture (Fig. 1A). The hESC-NP cultures stained uniformly for nestin (Fig. 1B) and Sox1 (Fig. 1C). Flow cytometry analysis revealed that 84.5 ± 4.5% and 79.0 ± 3.7% of the cells were positive for nestin (Fig. 1D) and Sox1 (Fig. 1E), respectively. Under our culture conditions, G-band karyotyping of these hESC-NPs confirmed a normal karyotype after 11 passages (Fig. 1F). Few hESC-NPs expressed neuronal markers such as MAP2 (Fig. 1G) and β-III-tubulin (TUJ1 staining; Fig. 1H), and the astroglial marker, GFAP (Fig. 1I).

The characterization of human embryonic stem cell-derived neural progenitors (hESC-NPs). Phase contrast photomicrograph of confluent hESC-NPs as a monolayer at passage 11 (A). Immunofluorescence staining indicated that the established hESC-NPs expressed nestin (B) and sex determining region Y-box1 (Sox1) (C). Flow cytometry analysis of hESC-NPs for the related markers (D, E). Cytogenetic evaluation of the hESC-NP line at passage 11 by standard G-banding (F). A few cells expressed neuronal microtubule associated protein 2 (MAP-2) (G) and β-III-tubulin (shown by Tuj1 staining) (H), and astroglial markers, glial fibrillary acidic protein (GFAP) (I). Quantitative real-time RT-PCR of hESC-NPs, relative to undifferentiated hESCs. The mean of each triplicate is plotted and error bars represent standard error (J). Growth factor removal from culture medium at passage 11 gave rise to spontaneous differentiation after 35 days. Phase contrast photomicrograph (K) and immunofluorescence staining for β-III-tubulin (TUJ1) (L), MAP2 (M), and GFAP markers (N). Scale bars: 200 μm (B, H, I), 100 μm (G). DAPI, 4′,6-diamidino-2-phenylindole; Otx2, orthodenticle homeobox 2; Hoxa2, homeobox a2; OCT3/4, octamer binding protein 3/4; PI, propidium iodide.
To assess the regional fate determination of hESC-NPs during rostrocaudal axis, the expression of region-specific transcription factors in hESC-NPs cultured for passage 20 was studied by qRT-PCR (Fig. 1J). The expression of pluripotent stemness markers, Oct4 and Nanog, and forebrain-rostral midbrain marker, Otx2, were downregulated. However, the expression of Hoxa2, Hoxa5, and Hoxb4, hindbrain markers, was upregulated, and Hoxc5, a cervical spinal cord marker was not changed. Therefore, it seems that the generated hESC-NPs are related to hindbrain.
We evaluated the potency of hESC-NPs to spontaneously undergo neural lineage differentiation by withdrawal of growth factors in differentiation (Fig. 1K). Immunofluorescence staining showed that our NPs could differentiate into neurons and glia (Fig. 1L—N).
Culture of SCs
SCs were isolated from adult sciatic nerve biopsies according to our established protocol, as described previously (60). SCs at passages 3—4 were used in this study. Purity of cells was above 99% as determined by flow cytometry. The viability after labeling with trypan blue showed more than 97% vitality of PKH-26-labeled cells. To evaluate whether or not permeabilization could fade away the membranous PKH-26 labeling, immunostaining for GFAP, a cytoplasm marker to assess SCs, was used (64). The results revealed that PKH had a stable attachment in the membrane and did not clean off through permeabilization with Triton X-100 from SCs (Fig. 2).

Isolated Schwann cells (SCs) in vitro. Phase-contrast micrograph of SCs (A). Immunostaining for p75 low-affinity nerve growth factor receptor (P75LNGFr) (B) and at higher magnification (C). Flow cytometry histogram for P75LNGFr (D). SC suspension immediately after labeling with PKH-26 (the red fluorescent cell membrane linker) labeled cells acquire a punctuate and/or patchy appearance (E), and 1 day after plating (F). Immunostaining of PKH labeled SCs for GFAP as a marker for glial cells (G—I). The stable attachment of the fluorescent dye into the membrane does not clean off through permeabilization with Triton X-100. Scale bars: 100 μm (A), 200 μm (B), 50 μm (C), 200 μm (E—I).
Coculture of hESC-NPs and SCs
To surmise the possible effects on SCs, NPs were cocultured with rat SCs in neural differentiation medium for 3 weeks (Fig. 3A). The adhesion of hESC-NPs in the coculture group was higher than the absence of SCs. Additionally, hESC-NPs formed a network with small windows when cocultured with SCs, and SCs were observed mainly within the windows of this network (Fig. 3B—D). Moreover, interestingly, neurons extended out longer processes when cultivated with SCs (Fig. 3D). Immunofluorescence staining of spontaneous differentiated hESC-NPs in the presence and absence of SCs revealed that cocultured hESC-NPs expressed neuronal markers to a significantly higher degree than those grown without SCs (TUJ1 staining: 74.8 ± 1.4 vs. 50.0 ± 0.9, p < 0.01; MAP2: 68.3 ± 1.8 vs. 45.4 ± 1.4, p < 0.01, Student's t-test) (Fig. 3E—H). The percentage of GFAP marker was lower in coculture when compared to the control (19 ± 1.7% vs. 32.0 ± 1.8%, p < 0.01, Student's t-test) (Fig. 3H). Together these data suggest that hESC-NPs differentiated preferentially to neuronal cells when cocultured on SCs.

Coculture of hESC-NPs and SCs. Schematic coculture illustration. Briefly, hESC-NPs were added on SCs. After 1 day, medium was switched into differentiation medium and continued for 3 weeks (A). Phase contrast photomicrograph of cocultivated NPs and SCs (B), and higher magnification in (C, D). Note the long processes that occur more often in a cocultivated system (D). Double immunostaining of cocultured hESC-NPs for HNu and β-III-tubulin (TUJ1) (E), or MAP2 (F) and/or GFAP (G). Quantitative immunofluorescence staining showed a significant increase in neuronal markers and decrease in GFAP expression in cocultured NPs in comparison with spontaneous differentiation of only hESC-NPs (H). The Student's t-test was used to compare immunostaining data between hESC-NP and hESC-NP/SC groups. a: p < 0.01. Scale bars: 200 μm (B), 100 μm (C, D), 200 μm (E—G).
Functional Recovery
Since the neuronal differentiation effect of SCs on NPs in vitro was seen, we hypothesized that cotransplantation of SCs with hESC-NPs could improve the functional recovery in contusion-injured spinal cords of rats by increasing neuronal differentiation in vivo. Using a dissecting microscope, a laminectomy was made at the T9-T10 spinal vertebrae, followed by a contusion injury at the T10 level in which a 10-g rod was dropped upon exposed vertebra. Figure 4A, B shows the spinal column before and after contusion. Hematoxylin and eosin (H&E) staining of coronal sections showed distinct interruption of myelin homogeneity and cavity formation that is usually observed in the contusion-injured spinal cord of rats (3) or primates (36) (Fig. 4C, D).
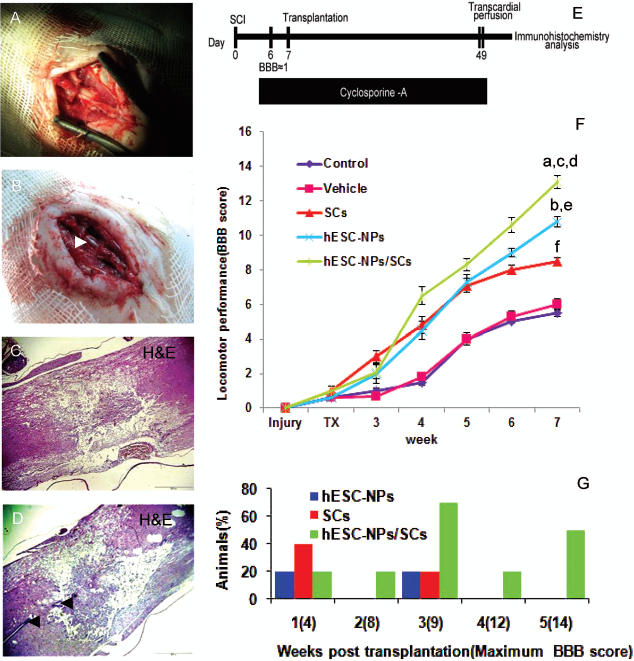
Injury, cell transplantation protocol, and Basso Beattie Bresnahan (BBB) score. Exposed spinal cord and surrounding meninges before contusion (A). Spinal cord after spinal cord injury (SCI); note the hemorrhage at the lesion spot (white arrow) (B). H&E-stained photomicrograph of lesion site representative of disintegration and interruption of spinal tissue (C). A section crossing the central canal of the contused spinal cord (black arrows) (D). Schematic illustration outlining in vivo procedure comprising study period, the time of injury, transplantation, cyclosporine administration (E). Hindlimb locomotor function assessment by BBB gait scale. TX indicates the time of transplantation. BBB score of hESC-NP/SC transplanted rats were significantly higher than control, vehicle, and individual cell grafts (F). Except for the first week after transplantation in which SC-transplanted animals displayed a better functional outcome, a larger proportion of hESC-NP/SC transplanted animals had a higher BBB score beginning from the second week until the study ended (G). We performed one-way ANOVA analysis followed by Tukey's post hoc test for multiple comparisons. n = 8 per group. a: p < 0.01, hESC-NP/SC group versus SC group; b: p < 0.01, hESC-NP group versus vehicle and control groups; c: p < 0.01, hESC-NP/SC group versus vehicle and control groups; d: p < 0.05, hESC-NP/SC group versus hESC-NP group; e: p < 0.05, hESC-NP group versus SC group; f: p < 0.05, SC group versus vehicle and control groups. Scale bars: 200 μm (C, D).
The timing of SCI, cyclosporine administration, and cell transplantation are outlined in Figure 4E. To evaluate the effect and fate of hESC-NPs and rat SCs in the SCI model, we transplanted the cells subacutely into the rat contused spinal cord (Fig. 4E). To determine locomotor recovery, BBB scoring of the open-field walking test was performed before the contusion and up to 6 weeks later (Fig. 4F). After SCI, animals displayed complete and flaccid paralysis of both hind limbs. To ensure that initial BBB scores were uniform between groups, BBB was tested at day 6 (1 day prior to transplantation). Rats with mean BBB scores of 1 were randomly included in our experimental groups. Open-field locomotion in the cell transplanted groups (hESC-NPs, hESC-NPs/SCs, and SCs) was significantly improved up to 5 weeks postimplantation compared to the vehicle and control groups (p < 0.01, one-way ANOVA, post hoc Tukey's test). Animals in the hESC-NP and SC groups showed a similar recovery for 3 weeks after transplantation. Afterwards, and by the end of the study, the BBB score of hESC-NPs increased (p < 0.05, one-way ANOVA, post hoc Tukey's test), while recovery of SC transplanted animals remained unchanged. At the fifth week posttransplantation, the highest functional recovery was observed in the hESC-NP/SC transplanted group in comparison with the hESC-NP or SC transplanted groups (at least p < 0.05, one-way ANOVA, post hoc Tukey's test). The hESC-NP/SC group achieved a mean BBB score of 13.1 ± 0.4. In contrast, the hESC-NP or SC transplanted groups only achieved mean BBB scores of 10.8 ± 0.3 and 8.5 ± 0.2, respectively. The BBB scores in hESC-NP or SC transplanted groups were significantly more in the vehicle (6.0 ± 0.3) and control groups (5.5 ± 0.2) (p < 0.01, one-way ANOVA, post hoc Tukey's test) (Fig. 4F).
We found that a larger percentage of animals had higher BBB scores in the hESC-NP/SC group compared to the hESC-NP or SC groups. For example, 50% of the rats that received hESC-NPs/SCs scored 14 compared to the hESC-NP or SC groups whose scores were less 5 weeks after transplantation (Fig. 4G).
Fate of Transplanted Cells
Animals were sacrificed after 5 weeks and immunocytochemistry was carried out on the cords to elucidate the possible mechanisms that led to the observed functional recovery. The survival of transplanted hESC-NPs and SCs was assessed using PKH labeling and a selective marker of human cell (HNu) immunostaining, respectively. Examination of the sections revealed that SCs were generally observed in the lesion cavity, HNu-positive cells were clearly visible around the lesion perimeter and migrated rostrocaudally to the surrounding gray and white matter (mainly caudal) (Fig. 5A, B). Sciatic nerve purified SCs expressed P75LNGFr marker, a specific cell surface maker. Examination of sections confirmed its expression on labeled SCs (Figs. 5E—H). Moreover, the presence of P75LNGFr-positive cells that were not PKH labeled or HNu positive, which indicated native migratory SCs to the injured spinal cord, was noted (6) (Fig. 5E—H). To determine the phenotype of HNu-positive cells, dual-label immunofluorescence was performed for neuronal, astroglial, oligodendroglial, mitotic, and progenitorial markers. To determine the percentage of cells expressing the respective markers, the numbers of HNu-positive cells were divided to their respective markers. The results disclosed that by the end of the fifth week of transplantation 12.3 ± 0.75%, 4.6 ± 0.25%, 15.3 ± 1.7%, and 31.1 ± 0.73% of cells were positive for TUJ1 staining, MAP2, myelin basic protein (MBP), and GFAP, respectively. Additionally, about one third of the engrafted cells (33.0 ± 0.77%) were nestin immunoreactive, which suggested that they remained largely undifferentiated. The presence of SCs in the hESC-NP/SC transplanted group was able to significantly boost the proportion of neuronal markers when compared with hESC-NP transplanted group (TUJ1 staining: 20.2 ± 0.5 in hESC-NPs/SCs vs. 12.3 ± 0.7 in hESC-NPs; MAP2: 12.0 ± 0.6 in hESC-NPs/SCs vs. 4.6 ± 0.2 in hESC-NPs, p < 0.01, Student's t-test) (Fig. 6). Furthermore, expression of nestin, a neural progenitor marker, was significantly reduced compared to the hESC-NP transplanted group (25.0 ± 0.6%.0 in hESC-NPs/SCs vs. 33.0 ± 0.7% in hESC-NPs, p < 0.01, Student's t-test) (Fig. 6). However, there was no significant difference in both transplanted groups for GFAP and MBP (GFAP: 29.4 ± 0.6 in hESC-NPs/SCs vs. 31.0 ± 0.7 in hESC-NPs, MBP: 17.0 ± 1.0 in hESC-NPs/SCs vs. 15.3 ± 1.2 in hESC-NPs) (Fig. 6). These results suggested that cotransplantation led to differentiation of hESC-NPs into more mature neurons compared to the hESC-NP group.

Tracing of transplanted cells in longitudinal sections of spinal cords. Low magnified (10x) image for transplanted hESC-NPs using human nuclei (HNu) antibody, 5 weeks after transplantation. hESC-NPs were mostly observed around the lesion cavity intermingled between spared white and gray matter. A few numbers of hESC-NPs were detected up to 8 mm away from the injury epicenter. The asterisk indicates lesion cavity (A). Higher magnification of boxed area in A (B). SCs survived and remained in the grafting site 5 weeks after transplantation (C). Longitudinal section of hESC-NP/SC transplanted spinal cord. PKH-26 indicates SCs mostly resided at the grafted area. HNu-positive staining indicates the extent of hESC-NPs that survived and migrated out of the lesion cavity and dispersed in a rostrocaudal direction (D). Microphotograph of PKH-26-labeled SCs for rat P75LNGFr demonstrates its expression on the plasmalemma of SCs (E-H). Moreover, the presence of PKH-26- and P75LNGFr+ cells indicates endogenous SC recruitment into the injured spinal cord. Scale bars: 2 mm (A), 1 mm (B—D), 200 μm (E-H).

The fate of hESC-NPs in the spinal cord. Representative figures related to cotransplanted hESC-NPs/SCs. Double immunostaining using HNu, hESC-NPs marker, and mestin, a progenitor marker (A); TUJ1, β-III-tubulin intermediated neuronal marker (B); MAP2, a mature neuron marker (C); GFAP, an astrocyte marker (D); myelin basic protein (MBP), a mature oligodendrocyte marker (E); and Ki67, a mitotic marker (F). Quantification of fate acquisition in hESC-NP and hESC-NP/SC groups (G). To compare immunostaining data between hESC-NP and hESC-NP/SC groups, Student's t-test was performed. a: p < 0.01. Scale bars: 200 μm (A, B, D, E, F), 500 μm (C).
Moreover, 4.0 ± 0.9% (in hESC-NP/SC group) and 4.6 ± 0.6% (in hESC-NP group) of the transplanted cells expressed Ki67, a cell cycle marker (Fig. 6). Ki67-positive cells were found randomly dispersed across the graft area without evidence of clustering in specific sites. There were no considerable differences in Ki67-positive cells between the cotransplant and hESC-NP groups (Fig. 6). No evidence of tumor formation was observed in the transplantation groups. Therefore, although teratomas are predominantly derived from the hESC graft, the purified hESC-derived differentiated cells provide an opportunity for cell-based therapies.
Discussion
Here we have shown SC transplantation 7 days after contusive injury at the 10th thoracic vertebral (T10) level resulted in significant locomotor functional recovery compared to the control and vehicle groups. SCs have been used following SCI, and it is known that SCs promote axonal regeneration and myelination after SCI (7,9,21). By this way, there was a significant reduction in cyst formation, increased tissue sparing, support of axonal growth into the graft, significant improvement in axon sparing and/or growth of spinal and supraspinal axons, and modest improvement in locomotor function (78). These improvements can be attributed to their membranes, extracellular matrix, and their production of diffusible trophic factors [for review see (87)]. Moreover, PKH labeling of the SCs has shown that the majority of the SCs remained in the grafted area, in accordance with previous studies (6,69,78). Modification of SCs to release neurotrophic factors (28,55) or the combination of SCs with other alternatives such as cAMP (69), OECs (68), and chondroitinase (26) may to a certain degree fulfill the endeavor appointed to SCs.
On the other hand, there are several reports that show transplantation of NPs or NSCs provides a promising future to improve functional recovery in the SCI rat model [for review see (79)]. NPs have been isolated from numerous regions in the developing and adult nervous system (19,89). Here, we have shown that NPs can generate in large quantities of enriched stable proliferating cells from hESCs using an adherent system and defined medium supplemented with a combination of inducing and growth factors. The differentiated cells highly expressed nestin and Sox1, molecular features of NPs, and had the caudal region identity. Our hESC-NPs have the capability of continuous self-renewal and normal karyotype in the presence of EGF and bFGF. They are also multipotent, as they give rise to neurons and glia after growth factor removal. The characteristics of our generated hESC-NPs provide the opportunity to exploit ready-to-use hESC-NPs for future biomedical applications. Recent studies have generated efficient NPs from hESCs that have self-renewal potential from rosette-forming cells isolated during early hESC differentiation stages (20,24,34,37,45,76).
There are limited data available on the transplantation of ESC-NPs in SCI (44,46) or severe sciatic nerve axotomy in rats (18). Recently, we have demonstrated that transplantation of hESC-derived neural tube-like structures, including primary NPs with the differentiation potential of neuronal and glial cells in collagen gels, improves functional recovery of SCI in adult rats subjected to midline lateral hemisection SCI (31). Therefore, in the present study we evaluated the therapeutic effects of transplanting self-renewable hESC-NPs on the functional recovery on a rat SCI contusion model. Interestingly, we found that transplantation of hESC-NPs improved significant locomotor functional recovery after SCI. This improvement was higher in hESC-NPs compared to SCs and exhibited neurogenic and gliogenic differentiation, which was similar to those seen in vitro. Moreover, the hESC-NPs in this study expressed MBP, which represented myelin-forming potential. Therefore, we hypothesize that our hESC-NPs were differentiated and integrated into spinal cord circuitry (e.g., forming new oligodendrocytes and/or neurons) (19,41). However, to confirm this it needs to abolish human engrafted cells by selective ablation with diphtheria toxin as determined previously (19). Our results are in agreement with previous reports showing that transplanted NPs can survive, migrate, and differentiate in vivo (13,33,67,70,85). The secretion of trophic factors is another possible mechanism (25,35,38,48,71,77). The possibility that hESC-NPs affect host locomotor recovery by means of multiple mechanisms such as increasing host neuronal, oligodendroglial, or axonal survival or decreasing host glial scaring, and increasing host-mediated regeneration or remyelination must also be considered.
To improve the neuronal and oligodendrocyte differentiation from transplanted hESC-NPs and generation of a proper microenvironment for treatment of acute SCI, here we have applied the cotransplantation of hESC-NPs with SCs. By this way, hESC-NPs leave the cavity into the spared tissue and emigrated both rostrocaudally. Moreover, immunostaining showed approximately twice the increment of TUJ1- and MAP2-positive cell formation compared to individual hESC-NP grafting 5 weeks after transplantation. Therefore, neuronal differentiation of hESC-NPs after cotransplantation into the lesioned spinal cord has been improved when compared with NP transplantation alone, in which fewer NPs differentiate into neuronal cells (13). This finding is of importance given that cotransplantation of SCs enhances more NP differentiation into neuronal cells. It seems that these promoting effects of the cotransplantation on hESC-NPs can be attributed to the properties of SCs, which have been previously discussed. Another aspect is that SC cotransplantation may drive the transplanted hESC-NPs to serve a more trophic role and alter the host microenvironment. Moreover, there is evidence that neurotrophins released by SCs could upregulate TrkC-mediated neuronal differentiation of NPs (11,14,92). The possibility of SCs in breaking down the myelin debris (83) and myelination prosperities could be another possibility in aforementioned recovery and suggest more specific evaluation on myelin characterization by electron microscopy (6,41). Alternatively, the mechanisms mediating recovery of function may be cell population interaction specific. Further studies are needed to determine the precise mechanism(s) of how SCs promote the survival and differentiation of hESC-NPs.
Moreover, we found several unlabeled SCs in the transplanted area, which suggest migration of endogenous SCs to the injury site after SCI despite being part of the peripheral nervous system (PNS) (4,10,32). This result shows that SC transplantation provides a microenvironment to increase neuronal differentiation of NPs and/or their survival, which does not exclude the potential contribution of endogenous SCs on transplanted hESC-NPs.
In summary, results from this study provide evidence that, compared with NPs and SCs alone, cotransplantation of hESC-NPs and SCs has displayed a higher functional improvement in rats with contused spinal cords. It may be that cotransplantation has, at least partially, optimized a microenvironment favoring survival and differentiation of cultured hESC-NPs transplanted into the lesioned site of the spinal cord. The next question would be whether the differentiated neuronal cells integrate into the spinal tissue and replace the damaged cells (19,89), or generate neural growth factors (40,51,53) that in turn promote local neurogenesis and axon outgrowth (33). Recently, the generation of patient-specific NPs from induced pluripotent stem cells (iPSCs) (59) provides an invaluable contribution to the field of regenerative medicine and improved locomotor function recovery in animal model of SCI (82). Therefore, the combination of results of our study with iPSC technology may shed new light onto the clinical application of pluripotent stem cell-derived NPs for the treatment of human SCI as well as other neuronal degenerative diseases.
Footnotes
Acknowledgments
This study was funded by a grant provided from Royan Institute and Iranian Council of Stem Cell Technology. The authors declare no conflicts of interest.