
Abstract
Duchenne muscular dystrophy (DMD) is the most frequent muscular dystrophy in children and young adults. Currently, there is no cure for the disease. The transplantation of healthy myoblasts is an experimental therapeutic strategy, since it could restore the expression of dystrophin in DMD muscles. Nevertheless, this cellular therapy is limited by immune reaction, low migration of the implanted cells, and high early cell death that could be at least partially due to anoikis. To avoid the lack of attachment of the cells to an extracellular matrix after the transplantation, which is the cause of anoikis, we tested the use of a fibrin gel for myoblast transplantation. In vitro, three concentrations of fibrinogen were compared (3, 20, and 50 mg/ml) to form a fibrin gel. A stiffer fibrin gel leads to less degradability and less proliferation of the cells. A concentration of 3 mg/ml fibrin gel enhanced the differentiation of the myoblasts earlier as a culture in monolayer. Human myoblasts were also transplanted in muscles of Rag/mdx mice in a fibrin gel or in a saline solution (control). The use of 3 mg/ml fibrin gel for cell transplantation increased not only the survival of the cells as measured after 5 days but also the number of fibers expressing dystrophin after 21 days, compared to the control. Moreover, the fibrin gel was also compared to a prosurvival cocktail. The survival of the myoblasts at 5 days was increased in both conditions compared to the control but the efficacy of the prosurvival cocktail was not significantly higher than the fibrin gel.
Introduction
Duchenne muscular dystrophy (DMD) is the most common genetic muscle disorder in children. The incidence is about 1/3,500 live male births and currently there is no efficient treatment (15). The cause of DMD is a severe deficiency of dystrophin, a major component of the dystrophin–glycoprotein complex in muscle fibers, responsible for the maintenance of sarcolemma integrity (10). Following the loss of a functional dystrophin protein, the muscles of DMD patients progressively degenerate as a result of continuous myofiber necrosis, leading to death into the second/third decade of the patient's life (17,31).
Cell transplantation offers hope for a future treatment of DMD. Different stem cell populations were proposed for transplantation such as satellite cells, myogenic progenitors, muscle-derived stem cells, and bone marrow stem cells (27,35). Satellite cells are muscle-committed stem cells that lie between the basement membrane and the sarcolemma. Satellite cells are quiescent, but under specific stimuli such as myofiber necrosis, some of them proliferate and differentiate as myoblasts, which subsequently activate myogenic differentiation to fuse among themselves to form new myofibers (17,33). These myoblasts can be proliferated in culture and transplanted in muscles to fuse together and form new fibers or to fuse with host myofibers, introducing in these hybrid fibers nuclei able to produce dystrophin in the case of DMD. Unfortunately, transplanted proliferating myogenic cells undergo rapid and massive cell death within a few days following implantation into skeletal muscles, as a result of cell dissociation, trophic factor withdrawal, oxidative stress, excitotoxicity, hypoxia, and possibly anoikis. Anoikis is a type of apoptosis that is triggered by detachment from the extracellular matrix and neighboring cells (18,20). Some studies pointed out that apoptosis, as well as anoikis, could be implicated in cell death after cell transplantation in muscles and in myocardium (13,21,32,34).
The goal of the present study is to test a biomaterial, which can serve as support for cell transplantation, providing a matrix around the cells to avoid anoikis during the first days after engraftment and that could disappear permitting the fusion of the cells with the surrounding myofibers. Despite the fact that natural hydrogels (i.e., collagen, Matrigel, fibrin) could present batch-to-batch variability, or some difficulties in chemical manipulation, they still appear to be superior to synthetic hydrogels for muscle tissue engineering, primarily due to the higher density of cell adhesion sites required for the 3D cell spreading (8).
Fibrin is a natural biomaterial used in tissue engineering resulting from the interaction of fibrinogen monomers and thrombin. Exposure of fibrinogen monomers to thrombin results in conversion of fibrinogen to fibrin, which polymerizes to form a fibrin mesh, by mechanisms similar to those involved in normal clotting in vivo (30). As a biological material, fibrin has some remarkable advantages over other synthetic materials: excellent physiological activities, selective cell adhesion, mechanical properties similar to natural tissues, and biodegradability. One advantage is its rapid degradation in vivo (1). Fibrin gel is a biomaterial, easy to form and use, controlled by the composition of fibrinogen and thrombin, and which could contain and progressively release growth factors. Moreover, fibrin gel can be injected and molded in situ to fill a tissular defect. The time of degradation can be increased by the presence of aprotinin in the fibrin gel (5,25). Moreover, some articles demonstrated that fibrin enhanced angiogenesis in the biomaterial, which is an important limiting factor in tissue engineering (2). Fibrin gel is still used in muscle tissue engineering. One study reported that a defect was created in a rat muscle and filled with myoblasts in a fibrin gel (5). The fibrin gel was formed in situ and muscle regeneration was observed after 12 weeks. Moreover, fibrin gel was also incorporated in a silicone chamber for the culture of cardiomyocytes for myocardial engineering (9). Huang et al. used fibrin gel to create an engineered self-organizing 3D skeletal muscle (24). This allowed the myoblasts to fuse into myotubes before the contraction of the gel. The gel can be maintained in culture 6 weeks. This model is interesting to understand the importance of electrical activity, hormonal signals and mechanical inputs for the comprehension of the development of skeletal muscle (24).
In the present study, the fibrin gel was used as a vector for human myoblast transplantation in mice, trying to avoid the cell death through anoikis. The cell survival was estimated with and without the fibrin gel and was compared to that obtained with a prosurvival cocktail (PSC). This PSC enhanced function of infarcted rat heart by improving graft survival of cardiomyocytes derived from human embryonic stem cells (28). The use of 3 mg/ml fibrin gel for cell transplantation enhanced both the survival of the transplanted cells by 30%, and also the number of myofibers expressing human dystrophin by 50% compared to the control muscles (myoblasts injected in saline).
Materials and Methods
In Vitro Studies
Reagents
Fetal bovine serum (FBS), Trizol, trypsin, and penicillin/streptomycin were obtained from Invitrogen (Burlington, Canada). Hank's balanced salt solution (HBSS), collagenase, XTT (2, 3-bis[2methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide inner salt) assay, phenazine methosulfate, human fibrinogen, human thrombin, phenol/chloroform/isoamyl alcohol, and sodium citrate were purchased from Sigma Aldrich (Oakville, Ontario, Canada). Proteinase K was purchased from Novagen (Gibbstown, NJ, USA). The [methyl-14C] thymidine was purchased from PerkinElmer (Boston, MA, USA). DNaseI, Superscript® Reverse Transcriptase, and DTT (dithiothreitol) were obtained from Invitrogen (Burlinton, Canada) and the random primers, Go Taq, oligo(dT) 15 primer, RNasin® from Promega (WI, USA). Aprotinin (Trasylol®) was purchased from Bayer Inc. (Toronto, Canada) and basic fibroblast growth factor (bFGF) from Gibco (Burlington, Canada).
Cell Culture
Myoblasts were grown from a muscle biopsy obtained from a 23-year-old man (a cadaveric organ donor) following informed consent of the family and approval by the Human Ethic Committee of the Centre Hospitalier de l'Université Laval's Research Center (CRCHUL). Human myoblasts were obtained after enzymatic digestion of the human muscle sample with collagenase (2%) in HBSS for 45 min at 37.5°C. The cells were then cultured at 37°C in a humidified atmosphere with 5% CO2 in proliferating medium MB1 (HyClone® MB-1 Medium, HyClone Laboratories, Inc., Logan, UT, USA) supplemented with 15% FBS, 10 ng/ml of bFGF, and 1% penicillin/streptomycin. Cells were used between passages 2 and 4 (6,12,14).
3D Culture in Fibrin Gel
Different solutions of human fibrinogen (3, 20, and 50 mg/ml) were made and mixed with dispersed cells at a density of 200,000 cells/ml, and a volume of 500 μl was placed in each well of a 24-well plate. Fibrinogen solution was mixed with human thrombin (50 U/ml) at a ratio of 0.03:1 (v/v) to fibrin. Culture medium was introduced on the top of the gel. Aprotinin (50 U/ml), an antifibrinolytic agent, was added in culture medium to maintain the fibrin gel integrity during the incubation period (19).
Comparison Between Monolayer, Fibrin Coating, and 3D Culture
Myoblasts were cultured in six-well plates for monolayer (on plastic) and fibrin coating. The fibrin coating was made by polymerizing fibrin gel (same conditions as below, 3 mg/ml fibrinogen) into the well before adding the cells. Then 100,000 cells were added into each well to be compared with the fibrin gel also containing 100,000 cells. The cells were cultured 7 days in MB1 supplemented with aprotinin.
Proliferation
XTT assay was performed according to the manufacturer's procedure (TOX2, Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada). Briefly, the cells were washed twice with HBSS. A mixture of 1 mg/ml XTT solution containing 1.5 mg/ml phenazine methosulfate solution was added to each well and incubated for 3 h at 37°C in an incubator with 5% CO2. Optical densities were then determined in a fluorometer (Spectra Max 340Pe molecular Devices, Sunnyvale, CA, USA) set at 450 nm absorbance and values were subtracted from the blank values. To estimate the number of cells present in the fibrin gel, a standard curve was made in the same conditions with increasing number of cells for each fibrin condition. The percentage of proliferation was then used to compare the different culture conditions.
Gene Expression
The total RNA was extracted using Trizol reagent from the myoblasts cultured on monolayer, on fibrin coating and into a fibrin gel. Firststrand cDNAs were synthesized using 1 μg total RNA with the oligo(dt) primer and Superscript III reverse transcriptase. Polymerase chain reaction (PCR) was performed with 1 μg of the cDNA solution using the primers specific for glyceraldehyde 3-phosphate dehydrogenase (GAPDH, as reference gene), myogenic differentiation (MyoD), myogenin, and myosin heavy chain 2 (MyHC). The annealing temperatures and the numbers of cycles of PCR are presented in the Table 1. PCR products were then separated by electrophoresis in 1% agarose gels and were stained with ethidium bromide. A picture of the gel was obtained with the GelDoc program (BioRad) under UV light.
Annealing Temperatures and Number of PCR Cycles

MyoD, myogenic differentiation; MyHC, myosin heavy chain 2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Degradation Time
Gels of 3, 20, and 50 mg/ml of fibrin containing 100,000 cells were made, as described above, in 24-well plates. The cells were cultured in medium without aprotinin. At different times (days 0, 1, 3, 5, and 7), the gels were recovered, put on an absorbing paper to eliminate residual medium, and then put into tubes to determine their wet weighs. The weight was expressed as a percentage of lost weight.
In Vivo Studies
Cell Transplantation
The transplantations were done in 2-month-old Rag/mdx mice produced in our animal facilities by crossing mdx/mdx mice (a DMD model due to a mutation leading to dystrophin deficiency) with >Rag–/– mice (an immunodeficient mouse that accepts human grafts). Mice were maintained under pathogen-free conditions. All the experiments with mice were conducted after approval by the CRCHUL Animal Care Committee and in agreement with the guidelines of the Canadian Council for Animal Care.
The tibialis anterior muscles (TA) of recipient mice were used for the implantation of 500,000 human male myoblasts per muscle. Briefly, the animals were under anesthesia with Isoflurane USP (Abbott Laboratories, Montreal, Canada). The skin was opened to expose the TA, and the myoblasts, resuspended in 10 ja.l of HBSS, were slowly injected obliquely using a glass micropipette (Drummond Scientific Co., Broomall, PA, USA) into 7–12 sites of the TA (7). The skin was closed with absorbable sutures.
Graft Conditions
Cells were resuspended in HBSS as previously described or in other conditions described below. In fibrin gel, the cells were mixed with the fibrinogen solution and a thrombin solution was added just before the graft to obtain a 3, 20, or 50 mg/ml fibrin gel in situ. A Pro-Survival Cocktail (PSC), described by Laflamme et al. in 2007 (28), was also tested. The PSC includes: 1) Matrigel to prevent anoikis, 2) a cell-permeant peptide from Bcl-XL (B-cell lymphoma-extra large) to block mitochondrial death pathways, 3) cyclosporine A to attenuate cyclophilin D-dependent mitochondrial pathways, 4) pinacidil, a compound that opens ATP-dependent K+ channels to mimic ischemic preconditioning, 5) insulin growth factor -1 (IGF-1) to activate Akt pathways, and 6) ZVAD-fmk, a caspase inhibitor. Matrigel was also tested alone.
Cell Death Assay at 5 Days
To quantify the in vivo cell mortality, myoblasts were radiolabeled by culturing them 48 h in growth medium containing 0.25 μCi/ml [methyl-14C]thymidine (50 mCi/mmol) before the transplantation. Radiolabeled cells (500,000 cells) were injected throughout the TA using a glass micropipette as previously described. The muscles were removed at days 0 and 5 after cell transplantation, snap frozen in liquid nitrogen, and stored at −80°C. DNA was extracted from transplanted TAs. Briefly, muscles were minced and incubated with 50 μl of proteinase K (10 mg/ml) at 56°C until the solution became clear. Digested muscles were then mixed with 500 μ l of a solution of phenol/chloroform/isoamyl alcohol (25:24:1) and centrifuged 3 min at 13,000 rpm. The upper solution was recovered and mixed with the same volume of chloroform and centrifuged again. The upper solution was recovered and 25 μl of 3 M citrate sodium was added before the addition of 1 ml of 100% ethanol. After centrifugation 8 min at 13,000 rpm, the pellets were washed in 70% alcohol before another centrifugation. The pellets were then dried before the suspension of the DNA in 100 μl sterile water. The amount of radiolabel within each TA was measured on DNA extracts using liquid scintillation counter (Mod. Wallac 1409, Woodbridge, Ontario, Canada). The primary data (dpm) were given for day 0 and day 5 as well as the amount of residual radioactivity at day 5 expressed as a percentage compared to day 0.
Human Dystrophin Detection (Graft Success at 21 Days Posttransplantation)
After transplantation of the cells in the different conditions mentioned above, the muscles were then recovered at 21 days after the surgery. Cryosections were made for an immunohistochemistry against the dystrophin. The sections were first washed with PBS. Nonspecific binding sites were blocked by incubating the cryostat sections with PBS containing 10% FBS, 10% goat serum, and 2% bovine serum albumin (BSA) for 30 min. Human dystrophin was detected with an anti-human dystrophin antibody (MANDYS104; MRIC Biochemistry Group, Wrexham, UK) (1:50 for 1 h), followed by a biotinylated anti-mouse antibody (DAKO, Copenhagen, Denmark) (1:200 for 30 min) and streptavidin-Cy3 (Sigma-Aldrich) (1:500 for 30 min). Sections were washed three times with PBS after each antibody incubation. The sections were blocked again for the second detection. Human nuclei were detected using mouse anti-human lamin A/C antibody (Vector Laboratory, Burlingame, CA) (1:100 for 1 h), followed by an anti-mouse Alexa 488 (Invitrogen) (1:300, 30 min). Dystrophin and lamin A/C staining were observed on PBS-glycerol-mounted slides under an ultraviolet lamp microscope using specific filters (510–560 and 450–490, respectively). The highest number of dystrophin-positive fibers was counted for each muscle. Three separate experiments of n = 5 were made for this study. To pool the three different experiments, results were expressed as a percentage of dystrophin-positive fibers compared to the number of dystrophin-positive fibers obtained in the control group (HBSS).
Statistical Analysis
Statistical significance was determined by Kruskal-Wallis chi-square test. Differences were considered to be significant at p ≤ 0.05 (*) and p ≤ 0.01 (**). The number of points or animals used for each study is indicated in the figure legends.
Results
In Vitro Studies
Effect of the Concentration of the Fibrin Gel on Myoblast Proliferation
Myoblast metabolism was determined with a XTT assay as an indicator of cell proliferation. The cells were cultured in different fibrin gel concentrations (i.e., 3, 20, and 50 mg/ml) during 7 days in the presence of aprotinin. The optical density (OD), corresponding to the metabolism of the cells at each time point and gel concentration, is shown in Figure 1A. The myoblasts cultured in the 3 mg/ml fibrin gel were metabolically the most active during all the culture time, followed by the cells into the 20 mg/ml fibrin gel and the cells with the lowest metabolic activity were those included in the 50 mg/ml fibrin gel.

Human myoblasts were cultured in fibrin gel at different concentrations of fibrinogen (3, 20, and 50 mg/ml). The metabolism of the cells (optical density values) into the gels was determined by a XTT (2, 3-bis[2methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide inner salt assay) (A). According to a standard curve, the number of cells was then estimated at 1, 3, and 7 days of culture and expressed as a percentage of cells relative to 100% at day 1 (B) (n ≥ 8; SD; *p ≤ 0.05).
The ODs were then converted as number of cells through a standard curve made for each fibrin condition. The results were expressed as a percentage of cells compared to day 1 (Fig. 1B). At day 3, the number of cells was not significantly increased compared to day 1. At 7 days, the number of cells increased 3.7 times in 3 mg/ml fibrin gel, 3.2 times in 20 mg/ml fibrin gel, and 2.5 times in the 50 mg/ml gel compared to the day 1. Moreover, the increases observed in the 3 and 20 mg/ml were significantly higher than that observed in 50 mg/ml fibrin gel. These results showed that the myoblasts proliferated more at lower fibrin densities.
Comparison Between Monolayer, Fibrin Coating, and Fibrin Gel
Myoblasts were cultured on plastic (monolayer condition), on a fibrin coating, or in a fibrin gel. Cell proliferation was quantified by XTT assay, as described above, after 7 days of culture in a medium containing aprotinin to maintain the gel. The results were expressed as a percentage of proliferation compared to day 1. After 7 days of culture, the number of cells increased by 8.7-fold in monolayer and on a fibrin coating (Fig. 2A). In a fibrin gel, the proliferation was significantly lower than in these two previous conditions, as the number of cells increased only by 3.7-fold.

Human myoblasts were cultured on plastic monolayer (ML), on fibrin coating (FM), and into a fibrin gel at 3 mg/ml (FG). After 7 days, cell proliferation was evaluated by XTT assay and reported as a percentage of proliferation compared to 100% at day 1 (A) (n ≥ 8; SD; *p ≤ 0.05). The state of differentiation of these cells was also examined by RT-PCR at day 7 (B). Different genes of differentiation were tested: myogenic differentiation (MyoD), myogenin, and myosin heavy chain (MyHC) and normalized to the expression of GAPDH (glyceraldehyde 3-phosphate dehyrogenase). M, molecular weight ladder.
Myoblast differentiation was also analyzed by RT-PCR and compared at day 7 between the three culture conditions (Fig. 2B). Myoblasts cultured on classic monolayer showed a higher MyoD expression compared to the cells cultured on fibrin or inside a fibrin gel. Moreover, in the presence of fibrin (coating or gel), the expression of myogenin was increased compared to the monolayer condition. MyHC expression was only observed in the fibrin gel correlating with the highest myogenin expression in that situation.
Time Rate of Degradation of the Different Fibrin Gels
Cells were cultured in the different fibrin gels under different concentrations of fibrinogen (3, 20, and 50 mg/ml) during 7 days and the wet weight of the gels with the included cells was measured every other day during 7 days of culture (Fig. 3). At day 3, only the 3 mg/ml gel had a weight significantly lower than the weight at day 0 and was significantly decreased compared to the two other gels. The 3 mg/ml fibrin gel continued to diminish at 5 and 7 days of culture, at which time only 5% of the initial weight was observed. The 20 and 50 mg/ml gels were the more stable, being significantly different from the day 0 weights only after 7 days of culture; the weights were then respectively 26% and 73% of the weight at day 0. Therefore, the 50 mg/ml gel was the most stable in culture. Indeed, the 3 mg/ml gel was almost completely degraded compared to the other two gels at day 7.

Human myoblasts were cultured in 3, 20, or 50 mg/ml fibrin gels. The wet weight of each gel was determined at 0, 1, 3, 5, and 7 days of culture and expressed as a percentage of weight loss from the day 0 (SD, n ≥ 6).
In Vivo Studies
Cell Survival 5 Days After Transplantation in Fibrin Gels of Different Concentrations
Radiolabeled cells were transplanted in mouse muscles that were recovered either at the transplantation day or 5 days later. The results were expressed as primary data (Fig. 4A) and as percentage of residual [14C]thymidine at 5 days compared to the day of the transplantation (Fig. 4B). These cells were grafted resuspended in HBSS or trapped in fibrin gels at different concentrations (3, 20, and 50 mg/ml). Only the 3 mg/ml fibrin gel was significantly higher than the control. A PSC has been previously reported to increase the survival of cardiomyocytes (28). This PSC contains Matrigel (Matrigel cocktail), and therefore Matrigel alone was also tested. When the cells were transplanted in HBSS, the percentage of residual 14C found after 5 days was only 5%. The Matrigel cocktail gave the highest percentage of residual radioactivity at day 5. Only two conditions were significantly different from the control, the fibrin at 3 mg/ml and the Matrigel cocktail, with 7.3% and 8.7% of radioactivity, respectively. However, there was no significant difference between these two groups. This experiment showed that using the fibrin gel alone increased myoblast survival as well as the PSC.

The death of the implanted cells was evaluated at 5 days posttransplantation. Human myoblasts were labeled with [14C]thymidine 48 h before their transplantation. Cells were resuspended in salin solution (HBSS) or in fibrin gels at 3, 20, or 50 mg/ml or in Matrigel or in a cocktail containing Matrigel (Matrigel cocktail) and transplanted in mouse tibialis anterior with a microsyringe. Muscles were taken at days 0 (J0) and 5 (J5). DNA extraction was made. The residual 14C-thymidine in the muscle was determined by scintigraphy and expressed as primarily data with the dpm values (A) or as a percentage of the value measured the transplantation day (B) (SEM; A and B: n ≥ 6; *p ≤ 0.05 different from the control).
Graft Success in Fibrin Gel
Several myofibers expressing human dystrophin were detected in the muscles recovered 21 days after human myoblast transplantation (Fig. 5A, B). The maximum number of dystrophin-positive fibers in a given complete cross section was determined for each muscle. Three experiments were made for this study and the number of dystrophin-positive fibers was reported for each experiment (Fig. 5C). For each experiment the average number of dystrophin-positive fibers was higher in the 3 mg/ml fibrin condition compared to the control. In order to have a global comparison of the results, the number of dystrophin-positive fibers in the control was expressed as 100% and the amount of dystrophin-positive fibers in the fibrin gel-grafted muscles was estimated compared to this control. We found a significant increase (54%) in the 3 mg/ml fibrin gel condition (Fig. 5D). The human nuclei were also observed in the same cryosections by fluorescent immunolabeling for human lamin A/C. This allows visualizing the location of the human nuclei relative to the human dystrophin-positive fibers. The human nuclei were all located into dystrophin-positive myofibers (results not shown). This observation confirmed that the transplanted cells had fused with the damaged myofibers. In the two groups, we observed dystrophin-positive fibers of different sizes. The small fibers could correspond to the fusion of transplanted cells exclusively among themselves to form new myofibers.
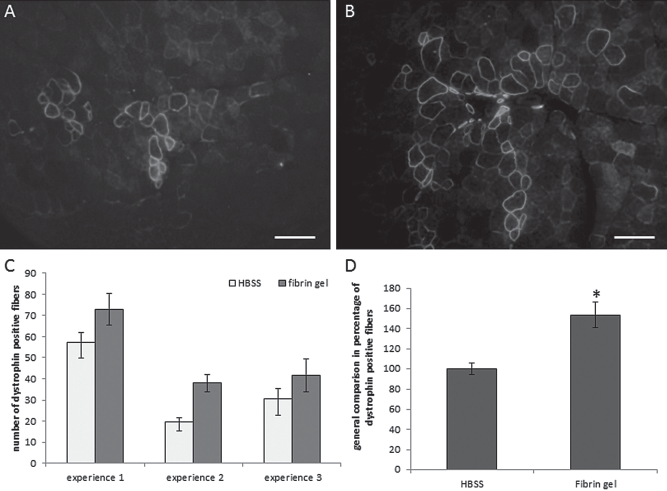
Human myoblasts were transplanted resuspended either in saline solution (HBSS) (A) or in fibrin gel (B) into the muscle of Rag/mdx mice. Muscles were recovered after 21 days. Immunohistofluorescence was used to detect human dystrophin in control (A) and fibrin gel (B) conditions (scale bars: 100 μm). The number of fibers positive for dystrophin was counted in each condition for the three different experiments (C: SEM; n ≥ 5) and a global comparison for the three experiments was made in percentage of dystrophin-positive fibers with the control reported to 100% (D, SEM; n ≥ 21; *p ≤ 0.05).
Discussion
In the present study, different concentrations of fibrin gel were used for the culture of human myoblasts. We observed that the stiffer the gel, the lower was the cell OD detected by a XTT assay. Indeed, the OD was different for each condition even at day 1. However, for each fibrin gel concentration, the OD increased at day 7 compared to day 1.
We also found that the stiffness of the gel influenced the cell proliferation. We found a 3.7-fold increase in the cell number between 1 and 7 days of culture in 3 mg/ml fibrin, and only a 2.5-fold increase for the 50 mg/ml fibrin gel. Our results with human primary myoblasts agree with those reported by Boontheekul et al. using a mouse myoblast line (11). Boontheekul et al. compared C2C12 and primary myoblasts in 3D alginate gel from different stiffness (11). They found that, when the C2C12 cells were seeded into the gel, their proliferation was increased in slowly degrading and stiffest gel. On the contrary, when the primary myoblasts were cultured in a stiffer gel, they proliferated less than in a degradable gel. They concluded that results obtained with the C2C12 cell line may not be predictive of the response of primary myoblasts to environmental cues. In another study, myoblasts were cultured 7 days in fibrin glue containing 100 mg/ml fibrinogen. These authors observed a great proliferation of the cells during the culture, which seemed to be higher than in our study for the 50 mg/ml fibrin gel. However, no estimation of proliferation was made and the myoblasts were cultured at higher concentration (200,000 cells in 150 μl of the fibrin glue) (16).
Our next step was to compare the culture of human myoblasts on plastic monolayer, on a fibrin gel or inside a fibrin gel. The proliferation of the cells was the same in both monolayer conditions, which was higher than in the 3D culture. In this last condition, the differentiation of the cells was earlier engaged. This process started with myoblasts, which are proliferative MyoD- and Myf5-positive cells. These proliferating myoblasts must withdraw from the cell cycle to become terminally differentiated myocytes that express late myogenic regulatory factors (MRFs), myogenin, and MRF4. The cells subsequently express muscle proteins such as MyHC and creatine kinase, and fuse to form multinucleated myofibers (11). The fibrin gel culture at 7 days not only showed the highest expression of myogenin but it was the only condition in which MyHC expression was detected. Similar results were observed by Bian et al., in a different culture condition than ours (i.e., 3% horse serum) (8). Fusion of myoblasts is frequently induced by changing the proliferating culture medium for a differentiation medium with no FBS or low percentage of FBS or horse serum (8,13).
Aprotinin is used in culture medium to prevent the degradation of the gel by the cells. The degradation process of the gel was observed in our study by culturing the cells without aprotinin and expressed as a gel weight loss compared to day 0. The more a gel is stiff, the less the cells will degrade it. At 7 days, the 50 mg/ml gel was still at 72% of the initial weight. It is known that as the concentration of fibrinogen or thrombin increases in a fibrin gel, its degradation becomes slow (25). In another study, cardiac cells were plated on a fibrin gel to form a bioengineered heart muscle. These authors added 200 μl of 20 mg/ml fibrinogen to a solution of 500 μl culture medium containing thrombin, which corresponds to a 5.7 mg/ml final gel (700 μl). One million cells were plated on this fibrin gel and they found that the gel was degraded within 2 weeks (23). The gel created by intracardiac injection of a mixture of 500,000 mesenchymal stem cells into 150 μl of fibrin glue at 20 mg/ml was still observed 10 days after injection in another group (30). Finally, 1 ml of a fibrin gel at 10 mg/ml containing 500,000 primary rat myoblasts was injected in a muscle defect. The presence of fibrin was still present 2 days after the transplantation but, 2 weeks later, the fibrin matrix was completely dissolved (5). The degradation rate was not studied in vivo in our experiments; the microinjections consisted of distributing 10 μl of the fibrin gel into 7–12 injection sites, which correspond to around 1 μl per site. We can only suppose that in this condition the degradation time would be very fast.
In our present study, the fibrin gel seems to be present in the muscle long enough to help reduce slightly the early cell death. The survival of transplanted cells was increased by 43% in a fibrin gel compared to the control. Christmann et al. used fibrin glue with a solution of 100 mg/ml fibrinogen. The commercial fibrin glue also contains aprotinin, plasminogen, plasmafibronectin, and factor VIII. They injected 5,000,000 myoblasts in 50 μl fibrin glue into infarcted myocardium. After 5 weeks, they found that the presence of fibrin glue increases angiogenesis and the grafted cell survival and doubled the percentage of covered area with the grafted myoblasts (16). Several approaches have been investigated to increase cell survival or long-term transplantation success, among them the introduction of vascular endothelial growth factor (VEGF) through electroporation of a plasmid (12), the pretreatment of cells with diazoxide or bFGF (22,26,29), or a coinjection of Matrigel containing bFGF (36). These are complex approaches leading to gains in survival ranging from 30% to 200% increases. Concerning the graft success, an increase from 30% to 400% was found in different experiments. In these studies, the cells were transfected with a lentivirus containing VEGF (12) or human follistatin (7), or pretreated with bFGF (26). In our case, only the cells transplanted in a fibrin gel, without any genetic modifications or growths factors supplementation, showed an increase in the number of positive fibers of more than 50%. Compared to a saline solution, the use of a biomaterial should improve the maintaining of the cells into the muscle, which would also explain the increased number of dystrophin-positive fibers. However, one article from Beauchamp et al. demonstrates that the incorporation of the donor cells within a fibrin clot did not prevent the loss of a majority of the transplanted myoblasts, compared to a saline solution, during the first 2 days after myoblast transplantation (4).
It is difficult to compare our results with those obtained by other teams because of the differences between the types of cells transplanted, including myoblasts from different species (human and mouse) and different ages (neonatal and adult cells). Moreover, the technique of transplantation may have a great impact in term of cell survival and graft success. Therefore, we cannot compare muscles electroporated, muscles treated with cardiotoxin, and muscle grafted using a glass capillary because the intensity of the damages created in the muscle is not comparable.
The intracardiac transplantation of cells in a PSC was used to enhance the function of infarcted rat hearts, although the authors did not estimate the cell survival after transplantation (28). The PSC contained different component including Matrigel. However, Matrigel cannot be used in clinical trials due to its murine origin and potential tumorigenicity (8). In our experiment, the PSC enhanced the cell survival compared to the control, but the improvement was not better than that obtained with the fibrin gel.
Finally, we want to emphasize that the percentage of residual [14C]thymidine radioactivity that we detected at posttransplantation day 5 does not necessarily correspond to the actual percentage of living cells. Previous studies demonstrated that, while many transplanted cells die, there is a surviving population that proliferates (3, 32). Since the radiolabel is lost by the dead cells and divided among proliferating cells, it can only reflect cell death but not cell proliferation (3,32). That means that there is probably more than 7% of living cells in the fibrin group. In spite of the fact that the reduction of the early cell death obtained with the fibrin gel is small from a clinical point of view, the present results are encouraging to progress in testing biomaterials and new prosurvival cocktails to improve the success of myoblast transplantation for potential future clinical applications.
Conclusion
Fibrin gel is a biocompatible material and has gained FDA approval for human use as sealant (with around 100 mg/ml fibrinogen solution). Unlike xenogenic gelatin or collagen, which may induce inflammatory and immune responses, fibrin gel can avoid the potential risk of a foreign body reaction when produced from the patient own blood. Fibrin gel could be easily implantable by injections through a syringe. This would obviate open surgery for treatment and therefore reduce the patient pain. Fibrin gel is also degraded enzymatically (25). Moreover, the use of fibrin gel in myoblast transplantation increased not only the survival of the transplanted cell at 5 days after the graft but also the number of dystrophin-positive fibers still present 21 days after the graft. Fibrin has the additional advantage that it binds growth factors, such as bFGF, which augment myogenesis (27). Therefore, it will be interesting to mix the fibrin gel with growth factors or other compounds to ameliorate the survival and graft success by releasing the active molecule during the degradation of the gel.
Footnotes
Acknowledgments
We thank Glenn E. Morris and Le Thanh Lam (MRIC Biochemistry Group, Wrexham, UK) for providing the MANDYS104 antibody. This work was supported by grants from the Jessee's Journey and from the Association Française contre les Myopathies (AFM). The authors declare no conflicts of interest.