
Abstract
Cell-based approaches using mesenchymal stromal precursor cells (MSCs) for the regeneration of intervertebral discs are attracting increased interest, even though the intervertebral disc is a very demanding environment. Implanted cells eventually face acidic pH, hypoxia, and a lack of nutrients. While the regenerative potential of MSCs for skeletal tissues has been well described, it is still questionable whether human MSCs can be prepared for prolonged survival and proper functioning and whether they can differentiate under the adverse conditions encountered in the disc. Here we examined the influence of hypoxia during expansion and differentiation on the chondrogenesis of MSCs. Chondrogenic differentiation was performed in in situ solidifying gelatin hydrogels, which represent a suitable matrix for delivering and anchoring cells within the disc tissue. To consider limitations in nutrition in the intervertebral disc, differentiation was performed at low cell concentrations in the gelatin hydrogels. Standard high-density micromass cultures served as reference controls. To determine the quality of chondrogenesis we analyzed typical marker molecules such as collagen types I, II, X, Sox-9, MIA, and aggrecan mRNA using RT-qPCR and determined protein deposition by histological stainings and biochemical methods. We could demonstrate that in gelatin-based hydrogels chondrogenic differentiation of human MSCs is possible at low cell concentrations. The quality of chondrogenic differentiation could be improved by hypoxia. Best results were obtained when the entire in vitro process, including MSC expansion and subsequent differentiation, was done under hypoxic conditions. MSCs that were expanded under reduced oxygen tension were primed for a chondrogenic differentiation.
Keywords
Introduction
The intervertebral disc is a demanding environment for cells. The nucleus pulposus is one of the most critical environments in our body, with an acidic pH, low oxygen tension (hypoxic conditions), and a paucity of basic nutrients (31,34,49). In addition, the cell concentration within the disc is low, making up only about 1% of the disc volume (24). These low cell numbers appear to be an adaptation to the environment, which has a limited supply of nutrients to support cell metabolism. Although this unsupportive environment poses challenges to successful tissue repair, cell-based approaches for disc regeneration, applying cell preparations from the removed prolaps tissue, are gaining interest (32). One reason is the high prevalence of degenerated disc disease in society and its socioeconomic consequences. Approximately 48.96 billion Euros in annual expenditures for direct and indirect back pain-related disorders have been estimated for Germany alone, amounting to 2.2% of the German gross domestic product (51).
In the case of a MRI-diagnosed degenerated disc in a patient with lower back pain, but without the need for prolaps surgery, mesenchymal stromal precursor cells (MSCs) have been considered for disc tissue engineering because autologous disc cells cannot be obtained without causing significant comorbidity to the donor disc (4). MSCs possess a high regenerative potential for skeletal tissues (10,18,21,35) and reveal a disc cell-like phenotype after in vitro chondrogenic differentiation in high-density micromass cultures or in alginate (39,46). They therefore seem to be a promising option for cell therapies of the degenerated disc. The feasibility of MSC-based therapies has already been shown in several animal models (5,8,14,15,41,45). However, it is still questionable whether MSCs can be prepared for prolonged survival and proper functioning under the above-mentioned adverse conditions.
Two strategies for the recruitment of MSCs for stem cell therapies are conceivable: first, the use of a purified population of freshly isolated MSCs (i.e., from the bone marrow) and their implantation into the disc without further in vitro manipulation. The second scenario employs in vitro expanded and/or in vitro differentiated MSCs. In this case, the MSCs can be preadapted to the limiting, hypoxic conditions in the disc. Environmental conditions such as oxygen tension, growth factors, and inflammatory agents can strongly influence growth and differentiation of MSCs (16,26,27). Adaptation under hypoxic conditions might therefore be a viable option. We recently presented evidence that, on the one hand, inflammatory processes inhibit the chondrogenic differentiation of MSCs, while on the other hand, hypoxic conditions exert a beneficial effect on chondrogenesis of MSCs (6).
Regardless of the chosen recruitment strategy for MSCs, a suitable implantation matrix is desirable to deliver and to anchor the cells within the tissue, preventing cell leakage from the injection site. In addition, after implantation the matrix could act to diminish the negative influence of the degenerated host tissue on the cells. Since healthy discs are highly hydrated and hyaluronan (HA)-rich tissues, HA-containing hydrogel matrices might be particularly suitable as a cell carrier.
Here we examined the influence of hypoxia both during expansion and during chondrogenic differentiation of mesenchymal progenitor cells on the success of the chondrogenic differentiation process. In addition to the high-density micromass culture (MM), chondrogenic differentiation was performed in in situ solidifying gelatin hydrogels with and without HA. To consider the limited nutrition in intervertebral discs, differentiation was performed at low cell concentrations in the gelatin hydrogels.
Materials and Methods
Isolation and Cultivation of Mesenchymal Stromal Precursor Cells (MSCs)
Bone marrow aspirates from patients undergoing total hip replacement were obtained from the BG Trauma Hospital Tübingen [n = 28; 13 female, mean age: 71 (range 57 to 89); 15 male, mean age: 60 (range 24 to 83)]. All procedures were approved by the local ethics committee. Informed consent was given for use of the MSCs for research. The mononuclear cell fraction was isolated using Ficoll-paque plus (GE Healthcare Europe GmbH, Freiburg, Germany) density gradient centrifugation for 30 min at 400 x g. Isolated cells were seeded into cell culture flasks. Adherent cells (MSCs) were expanded in expansion medium (42) consisting of 60% low-glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Karlsruhe, Germany), 40% MCDB 201 (Sigma-Aldrich, Steinheim, Germany), penicillin/streptomycin (Invitrogen), 0.05 μM dexamethasone, ITS+1 supplement, 0.1 mM l-ascorbic acid 2-phosphate (all from Sigma-Aldrich), 2% fetal bovine serum (Biochrom AG, Berlin, Germany), 10 ng/ml platelet-derived growth factor BB, and 10 ng/ml epidermal growth factor (both from Miltenyi Biotech, Bergisch Gladbach, Germany). When MSCs reached approximately 80% confluence, they were detached with 0.25% trypsin/EDTA (PAA Laboratories GmbH, Pasching, Germany) and stored in liquid nitrogen. Thawed cells were expanded for one additional passage before they were used for chondrogenic differentiation experiments.
Induction of Chondrogenic Differentiation
MSCs were differentiated chondrogenically as MMs and in hydrogels. Micromasses were produced by pipetting 20 μl of the MSC suspension, containing 0.4 million cells, into an uncoated well of a 96-well plate (17,37). Medium was added to the cells after 2 h. Three days later the generated MM was transferred into a well of a 12-well plate. In order to achieve chondrogenic differentiation in hydrogels, 0.4 million MSCs were embedded in 500 μl gelatin-based hydrogel matrices. Gelatin hydrogels containing 10% gelatin from pig skin (provided by Gelita AG, Eberbach, Germany) and gelatin-hyaluronan hydrogel containing 10% gelatin and 3.5 mg/ml HA (Visiol, TRB Chemedica AG, Haar, Germany) were used. The gelatin was enzymatically cross-linked by adding 0.15 U transglutaminase. Polymerization time was approximately 30 min.
MSCs were differentiated in chondrogenic differentiation medium (42) consisting of high-glucose DMEM, 1 mM sodium pyruvate, penicillin/streptomycin (all from Invitrogen), 0.1 μM dexamethasone, ITS+1 supplement, 0.17 mM l-ascorbic acid 2-phosphate, 0.35 mM l-proline (all from Sigma-Aldrich), and 10 ng/ml transforming growth factor-β3 (Miltenyi Biotech). The medium was renewed twice a week. For the hypoxia experiments the medium was renewed once a week to minimize unintentional gas exchange effects.
Gene expression was analyzed for at least 2 weeks in chondrogenically differentiated MSCs (mRNA expression remained stable after 2 weeks); protein expression was analyzed in MSCs differentiated for 8 and 9 weeks, respectively.
Establishing Hypoxic Conditions and Experimental Setup
Both MSC expansion and chondrogenic differentiation were performed under standard normoxic conditions (ambient oxygen tension of 20%) as well as under hypoxic conditions. Bone marrow in general is hypoxic, where some regions are as low as ~1–2% O2 [for review see (26)]. We defined an oxygen tension of 4% as hypoxia. A gas mixture of 5% carbon dioxide and 95% nitrogen was used to create the hypoxic atmosphere.
The experimental setup for hypoxia experiments is shown in Figure 1. MSCs were expanded normoxically until they reached about 80% confluence. After passaging, the MSCs were further expanded for 7 to 13 days, one part under normoxic conditions and the other part under hypoxic conditions. After detaching the proliferated MSCs, the cells were differentiated chondrogenically under hypoxia and under normoxia.
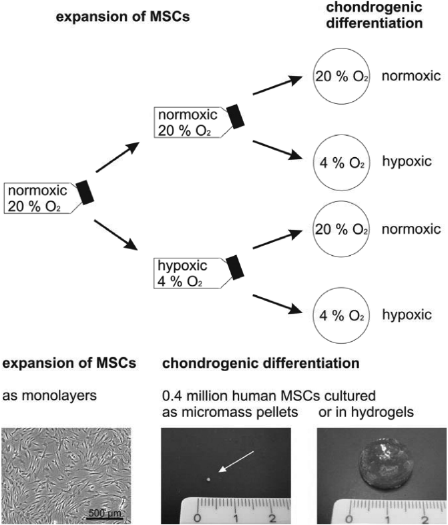
Experimental setup for hypoxia experiments.
Measurement of the Size of Micromass Pellets
The size of MMs was measured 2 weeks after differentiation of MSCs. Bright field microscopical images were taken and the cross-sectional areas were measured using AxioVision software (Carl Zeiss MicroImaging GmbH, Jena, Germany).
mRNA Isolation and Reverse Transcription
Hydrogels with chondrogenically differentiated MSCs were digested with 4 mg/ml collagenase B (Roche, Penzberg, Germany) at 37°C. After centrifugation the cell pellet was lyzed in Buffer RLT (Qiagen, Hilden, Germany). MMs were ground with Molecular Grinding Resin from G-Biosciences (Maryland Heights, USA) and directly lyzed in Buffer RLT. MSC monolayers serving as negative control were detached with 0.25% trypsin/EDTA. The cell suspension was centrifuged and the cell pellet was lyzed in Buffer RLT. RNA was isolated with the RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. An on-column DNase digestion step with the RNase-Free DNase Set (Qiagen) was added during RNA purification. The Reverse Transcriptase Core Kit from Eurogentec (Cologne, Germany) with oligo d(T)15VN primers was used for reverse transcription (RT) according to the manufacturer's recommendations. A maximum of 1 μg mRNA was transcribed into cDNA.
RT-qPCR
For quantitative PCR (qPCR) it is necessary to have appropriate reference genes that show no expression variations when exposed to different culture conditions. In this study we examined MSCs cultivated under a variety of culture conditions. They were cultured in monolayers as well as in three-dimensional cultures, were differentiated chondrogenically, and they were exposed to varying oxygen conditions. To determine an appropriate reference gene that is stably expressed under this variety of conditions, we bioinformatically analyzed the expression of five genes [tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta (YWHAZ), glyceraldehyde 3-phosphate dehydrogenase (GAPDH), hypoxanthine phosphoribosyl transferase 1 (HPRT I), ubiquitin C (UBC), and β-actin]. The analyses were performed with geNorm software (http://medgen.ugent.be/genorm) (50). YWHAZ (50) showed the fewest changes in its expression under all tested conditions (data not shown). For this reason we selected YWHAZ for use as the reference gene in this study. Our results were confirmed by Fink and colleagues, who also identified YWHAZ as the appropriate reference gene for the differentiation of human adipose-derived stem cells under hypoxic conditions (7). The qPCR MasterMix Plus w/o UNG for SYBR Assay Low ROX (Eurogentec, Cologne, Germany) was used for reverse transcription quantitative polymerase chain reaction (RT-qPCR). The PCR was run and analyzed on an Applied Biosystems 7500 Fast Real Time PCR System with 7500 Fast System SDS Software (Applied Biosystems Inc., Foster City, USA). mRNA expression levels were analyzed for collagen type I (COL1A2), collagen type II (COL2A1), aggrecan (ACAN), melanoma inhibitory activity (MIA), sex determining region Y-box 9 (Sox-9), and collagen type X (COL10A1). Primers were used at 300 nM (Table 1). The specificity of all primers was confirmed via BLAST analysis (1). PCR efficiencies (E) were calculated for each primer pair using a calibration curve. Quantification cycles (Cq) were determined for each gene. Relative gene expression was calculated according to the following equation: relative mRNA expression = [ECq(reference gene)]/[ECq(marker gene)]. Relative COL2A1 mRNA expressions in MSC monolayers were defined as 10−7 when no amplification signal was detectable. The influence of hypoxic culture conditions on chondrogenic differentiation was evaluated by recording the x-fold increase in relative mRNA expression in comparison to relative mRNA expression levels of standard normoxic cultures. A change less than twofold was defined as no change.
Sequences and Efficiencies of Primers for RT-qPCR Analysis

Sulfated Glycosaminoglycan (sGAG) Quantification
To determine the amount of deposited sGAG within the gel after 8 weeks of differentiation, hydrogels were digested with 1 ml of 4 mg/ml collagenase B. After 8 weeks of chondrogenic differentiation MMs were digested in 1 mg/ml papain (Sigma-Aldrich) prior to sGAG quantification. 1,9-Dimethylmethylene blue (SERVA Electrophoresis GmbH, Heidelberg, Germany) was added to the samples and absorbance was determined at 520 nm with the PHERAstar photometer (BMG Labtech GmbH, Offenburg, Germany). The concentration of sGAG was extrapolated from a chondroitin sulfate (Sigma-Aldrich) standard curve with a range from 5 to 200 μg/ml. The standards were prepared in collagenase-digested hydrogels without cells or in papain solution. A detection limit of 7 μg/ml was determined for gelatin hydrogels.
Histochemical Stainings
To visualize living cells within gelatin hydrogels after 9 weeks of chondrogenic differentiation, calcein AM (Invitrogen) was added to the cultures for 30 min. Stained gels were gently squeezed between the glass slide and the cover slip and three-dimensional images were acquired using the ApoTome system from Zeiss and a monochromatic camera (AxioCam MRm; Carl Zeiss MicroImaging GmbH).
Cryosections were prepared for histological examination. Cultivated MMs, gelatin hydrogels with and without HA, and a piece of a human nucleus pulposus obtained from prolaps surgery (provided by BG Trauma Hospital Tubingen) were embedded in Tissue-Tek O.C.T. Compound (Sakura Finetek Germany GmbH, Staufen, Germany), and the MMs and disc tissue were cut into 10-μm slices; hydrogels were cut into 50-μm slices. To examine the distribution of the differentiated cells within the hydrogels after 2 weeks in culture the cell nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole; Sigma-Aldrich).
To visualize sGAG of the extracellular matrix a selective Alcian blue staining was performed on micromass pellet slices and on gelatin hydrogel slices (after 8 weeks of chondrogenic differentiation). To minimize background staining due to gelatin contaminants, keratan sulfates were stained selectively with Alcian blue (Sigma-Aldrich) at pH 5.8 in the presence of 0.8 M magnesium chloride (Sigma-Aldrich) (44). Staining was examined with bright field microscopy (Axiophot) and a color camera (AxioCam MRc5; both Carl Zeiss MicroImaging GmbH).
Immunological staining for collagen type I, aggrecan, and collagen type X was performed using mouse antihuman antibodies. For collagen type I staining clone I-8H5 was used (20 μg/ml; MP Biomedicals, Illkirch, France), for aggrecan staining clone HAG7D4 was used (1:40; Acris Antibodies GmbH, Herford, Germany) and for collagen type X staining an antibody produced by the group of Klaus von der Mark was used (9). Collagen type II staining was performed using a mouse antichicken collagen type II antibody (clone II-II6B3; 1 μg/ ml; Linsenmayer) (25). The collagen type II antibody was obtained from the Developmental Studies Hybridoma Bank maintained by the Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine (Baltimore, MD), and the Department of Biological Sciences, University of Iowa (Iowa City, IA), under contract NO1-HD-2-3144 from the NICHD. The staining was visualized using a Cy3-coupled secondary IgG antibody from goat (3 μg/ml; Dianova, Hamburg, Germany). Sections were examined with a Zeiss Axiovert 200 M inverted microscope.
Statistical Analyses
Statistical analyses were performed with StatView (SAS Institute Inc.). p-Values were determined with t-test when two normally distributed groups with equal variances were compared. For multiple comparisons analysis of variance (ANOVA) was performed followed by Bonferroni post hoc test. Data sets were logarithmized for ANOVA analyses. Values lower than p < 0.05 were considered statistically significant.
Results
Human Bone Marrow-Derived MSCs Can Be Differentiated Chondrogenically at Low Cell Densities in Gelatin-Based Hydrogels
We analyzed the capability of human bone marrow-derived MSCs to differentiate chondrogenically at low cell densities in gelatin-based hydrogels. Standard carrier-free MMs served as control cultures to assess differentiation capacity. Both types of culture models, MMs and hydrogels, are three-dimensional differentiation cultures, but show strong differences in size and cell distribution. The differences in appearance are shown in the photographs of Figure 1. In MMs the cells are very densely packed, as revealed by DAPI staining in Figure 2A. In contrast, within the hydrogels the cells are widely spaced (Fig. 2B). The distribution and cell numbers are comparable to those of the nucleus pulposus tissue of the intervertebral disc (Fig. 2C). We embedded MSCs in gelatin-based hydrogels in concentrations of 0.4 million cells per 500 μl hydrogel to mimic in vivo conditions. During culture no shrinkage of the gels was observed (data not shown). To identify living cells within the gels during culture we performed calcein AM staining. Figure 2D shows that MSCs within gelatin hydrogels were still viable after 9 weeks of differentiation. No differences in cell distribution and viability of MSCs were observed between gelatin and gelatin-HA hydrogels (data not shown).

Cell distribution in micromass pellet (A), gelatin hydrogel (B), and native disc tissue (nucleus pulposus; C). MSCs were differentiated for 2 weeks as micromass pellets and in gelatin hydrogels. After fixation, cryosections were prepared and stained with DAPI. Cell distribution was compared to a cryosection of native nucleus pulposus tissue. The viability of differentiated cells in gelatin hydrogels (D) is shown after 9 weeks of differentiation using calcein AM. A three-dimensional reconstitution was performed after capturing images with the ApoTome from Zeiss.
To examine whether MSCs are able to differentiate chondrogenically within gelatin-based hydrogels, we analyzed the mRNA expression of marker genes via RT-qPCR. Expression levels for COL1A2, COL2A1, ACAN, MIA, Sox-9, and COL10A1 were measured and normalized to the reference gene YWHAZ (Fig. 3A). Successful chondrogenic differentiation was confirmed by the induction of the marker genes. Although there were huge donor-to-donor variations in the expression of all tested genes, their expression levels were significantly higher in chondrogenically differentiated MSCs than in nondifferentiated ones. Chondrogenesis could be induced successfully in MM and in hydrogels (Fig. 3A). COL1A2 expression was increased 5- to 13-fold in MM and in both gelatin-based hydrogels, COL2A1 expression 2 × 102- to 9 × 103-fold, ACAN expression 4 × 102- to 3 × 103-fold, MIA expression 5 × 102- to 2 × 103-fold, Sox-9 expression 14- to 34-fold, and COL10A1 expression 7 × 103- to 6 × 104-fold (median values).

(A) mRNA expression pattern of undifferentiated and chondrogenically differentiated human bone marrow-derived MSCs. The mRNA expression of collagen type I (COL1A2), collagen type II (COL2A1), aggrecan (ACAN), MIA, Sox-9, and collagen type X (COL10A1) was normalized to the reference gene tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide (YWHAZ). The expression levels of nondifferentiated MSCs (MSC) and differentiated MSCs from each donor are shown together with median expression levels. MSCs were differentiated as micromasses (MM), in gelatin (G), and in gelatin-hyaluronan (GH) hydrogels for 4 weeks. Black stars mark statistically significant differences between undifferentiated and differentiated MSCs, while gray stars refer to significant differences between differentiated samples (⋆⋆p < 0.01; ⋆⋆⋆p < 0.001, ANOVA-Bonferroni). (B) Expression of collagen types I, II, X and of aggrecan is shown by histochemical stainings of gelatin hydrogel slices with embedded MSCs. Chondrogenesis was induced for 9 weeks.
Comparisons of micromasses and hydrogel cultures revealed only minor differences in marker gene expression. COL1A2 mRNA expression was slightly but significantly increased in gelatin hydrogels (median expression: 76) compared to micromasses (median expression: 29). The differentiation pattern of MSCs in gelatin was comparable to that of MSCs differentiated in gelatin-hyaluronan hydrogels. The expression levels of all tested genes did not differ significantly between the two hydrogel types. But in general the expression level of all examined genes tended to be lower in HA-containing gelatins (nonsignificant). The expression of collagen type I, collagen type II, aggrecan, and collagen type X on the protein level could be shown by immunohistochemical stainings. Figure 3B shows the deposition of the molecules within gelatin hydrogels after 9 weeks of chondrogenic differentiation.
The Reduction of Oxygen to 4% During Culture Improves Chondrogenic Capability of MSCs
We wanted to analyze the effects of reduced oxygen tension (4% oxygen) on chondrogenic differentiation of MSCs within gelatin-based hydrogel cultures. Our experimental setup was designed in such a way that we could examine the effects of oxygen during MSC expansion and differentiation separately (Fig. 1) by first dividing normoxically expanded MSCs and subsequently expanding the two populations either normoxically or hypoxically. These expansion cultures were trypsinated and were embedded in hydrogels or cultured as MMs, and then differentiated at 4% oxygen or 20% oxygen.
During culture it became apparent that the size of the micromass pellets varied under the influence of oxygen tension (Fig. 4). MMs produced with hypoxically expanded MSCs had significantly larger cross-sectional areas (1.0 mm2) than those produced with normoxically expanded MSCs (0.8 and 0.7 mm2). There were two outliers from the same donor with an area of more than 2.5 mm2. Even after excluding those outliers from statistical analysis we found a significant effect. In contrast, no significant effects on MM size were observable when the size of hypoxically differentiated MMs was compared with that of normoxically differentiated MMs.

Cross-sectional area of micromass pellets. MSCs were cultured as described in Figure 1. After 2 weeks of differentiation the size of the MMs was measured. The figure shows MM size in mm2 for each donor and median values. +p < 0.05, t-test.
The effect of reduced oxygen tension on MSCs differentiated in gelatin-based hydrogels was examined by RT-qPCR as well as by sGAG quantification. Marker gene mRNA expression was analyzed in cells differentiated for 2 and 8 weeks. We determined that the effects of hypoxia on mRNA expression did not change over time, so the results of both time points are not shown separately. We compared gene expressions of COL2A1, ACAN, MIA, Sox-9, COL1A2, and COL10A1 after hypoxic MSC expansion and/or differentiation with those after standard cultivation (normoxic ambient oxygen tension) to evaluate the effect of hypoxia (Fig. 5).
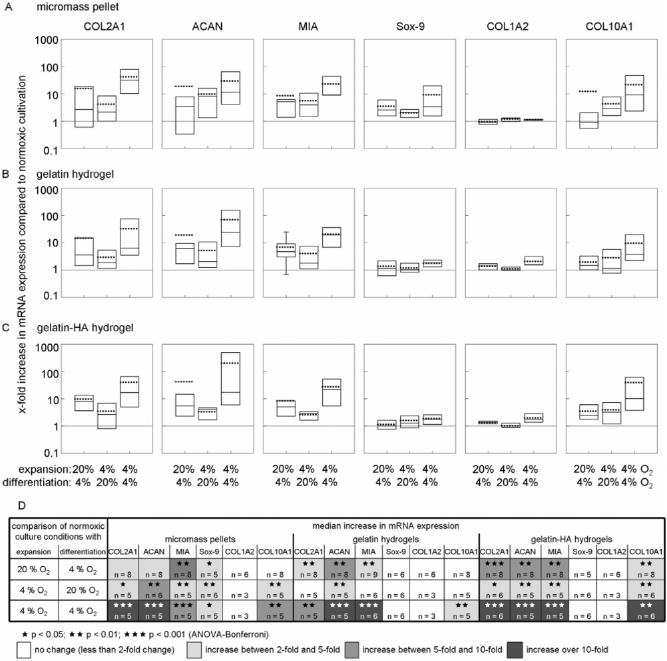
Effect of hypoxia on mRNA expression of chondrogenic markers in differentiated MSCs. The influence of hypoxia (4% O2) on micromass pellets (A), gelatin hydrogels (B), and gelatin-HA hydrogels (C) is shown; changes in marker gene expression are shown as x-fold changes compared to standard cultures (expansion and differentiation at 20% O2). The 25th and 75th percentile values are shown as boxes, and whiskers indicate the 10th and 90th percentiles. The line inside the box marks the median, and the dotted line represents the mean value. Significant differences (stars) and the number of tested donors for each group and gene are shown in (D).
Differentiation of MSCs under 4% oxygen tension after normoxic expansion (first bars in diagrams in Fig. 5A–C and first row in Fig. 5D) led to significant increases in COL2A1, ACAN, and MIA expression in both hydrogels. An ACAN increase of more than fivefold in median could be determined in both hydrogels. The median expression levels of COL2A1 and MIA increased in gelatin-HA hydrogels more (>5-fold) than in gelatin gels (>3-fold) compared to the standard conditions (normoxic expansion/normoxic differentiation). In addition, the expression of Col10A1 increased significantly in gelatin-HA hydrogels (2.4-fold), whereas in gelatin hydrogels no increase was detected. External treatment with hypoxia during the differentiation process was less effective on marker gene expression in MM, only the expressions of MIA and Sox-9 were increased significantly (median 5.2-fold and 2.6-fold).
The expansion of MSCs under hypoxic conditions did not enhance chondrogenesis in gelatin hydrogels, when the subsequent differentiation was carried out under normoxic conditions (second bars in diagrams in Fig. 5A–C and second row in Fig. 5D). Only mRNA expression of ACAN was found to be increased significantly (median 2.1-fold). In gelatin-HA hydrogels an increased expression could be detected for COL2A1 (2.6fold), ACAN (4.2-fold), MIA (3.0-fold), and COL10A1 (3.3-fold). In MMs the effects were similar to those of gelatin-HA hydrogels. The median increase was 2.2-fold for COL2A1, 8.2-fold for ACAN, 4.0-fold for MIA, 2.1fold for Sox-9, and 2.9-fold for COL10A1.
Hypoxia was most effective when applied during the whole culture process (expansion plus differentiation). This was true for MMs and for hydrogels, regardless of the presence or absence of HA (last bars in diagrams in Fig. 5A–C and third row in Fig. 5D). MSCs cultured in this way in gelatin hydrogels produced more than five times as much COL2A1, about 20 times as much ACAN (24.2-fold) and MIA (19.6-fold), and more than three times as much COL10A1 mRNA as cultures done completely at 20% oxygen tension (in median). A more pronounced effect could be found for gelatin-HA hydrogels. More than 10 times as much COL2A1 (16.9-fold), ACAN (17.3-fold), MIA (21.6-fold), and COL10A1 (10.3-fold) as after normoxic cultivation could be detected if the cells were cultured under hypoxia. In MMs mRNA expression of COL2A1 and ACAN was increased more than 10-fold (31.7-fold and 11.6-fold), MIA and COL10A1 more than 9-fold, and Sox-9 more than 3-fold (in median).
The expression of COL1A2 was not altered significantly by oxygen tension in any of our experiments. No alteration of Sox-9 expression was detectable in hydrogels.
To confirm the results of our gene expression analysis we examined the deposition of extracellular matrix at the protein level. The deposition of keratan sulfates was examined via selective Alcian blue staining. It showed the homogenous occurrence of keratan sulfates in MM as well as in gelatin hydrogels in all cultures, regardless of oxygen conditions (Fig. 6A). In gelatin gels the cells were distributed sparsely (Figs. 1 and 2B), so the staining appears to be less intense than in micromass pellets. For this reason no differences can be seen when the cells are cultured under hypoxic conditions.

Deposition of extracellular matrix within micromass pellets and gelatin hydrogels after differentiation of MSCs for 8 weeks. (A) Keratan sulfates of the extracellular matrix were stained with Alcian blue. (B) The amount of deposited sulfated glycosaminoglycan (sGAG) was determined in μg/0.4 million of initially differentiated MSCs using 1,9-dimethylmethylene blue.
To acquire a quantitative figure for the amount of deposited extracellular matrix we performed sGAG quantification using 1,9-dimethylmethylene blue. sGAG, which was deposited within 8 weeks in gelatin hydrogels and in MMs, was quantified for three donors (Fig. 6B). There were huge variances from less than 7 μg sGAG per gelatin hydrogel (0.4 million embedded MSCs) to more than 100 μg sGAG.
Analysis revealed that sGAG increased when expansion of MSCs was performed at 4% oxygen tension. The amount of sGAG in hydrogels was less than 7 μg for two of the three donors when MSCs were expanded normoxically. Hypoxic expansion led to sGAG deposition of 11 μg in the gelatin gel of one donor and 7 and 21 μg in those of the second donor. In gelatin hydrogels of the third donor the amount of sGAG was increased threefold through hypoxic MSC expansion. Normoxic expansion led to a mean deposition of 38 μg sGAG, compared to 118 μg after hypoxic expansion.
Increased sGAG deposition could also be detected in MMs. The MM from MSCs of the third donor contained 15 μg sGAG when cultured normoxically, increasing to 52 μg when differentiation was performed under hypoxic conditions. A mean of 155 μg sGAG could be detected in MMs when MSCs were expanded with 4% oxygen tension prior to chondrogenic differentiation.
Our results show that oxygen tension during MSC expansion effects chondrogenic differentiation more efficiently than during differentiation, regardless of whether hydrogels or MMs were used for differentiation.
Discussion
A combination of gelatin hydrogels and hypoxic conditions seems to represent a conducive environment for chondrogenic differentiation of human mesenchymal stromal precursor cells. After induction of chondrogenesis with TGF-β3, MSCs embedded in gelatin hydrogels remained viable for at least 9 weeks, developing a differentiation pattern similar to that of cells differentiated in MMs. MMs are most frequently used as a standard model for in vitro differentiation of MSCs into the chondrogenic lineage (17,37). However, they are not suitable for clinical applications due to their dense, solid structure, which virtually precludes the dissemination of single cells into a therapeutic environment. The gelatin hydrogels presented here as a model for the preparation of chondrogenically differentiated MSCs would allow such dispersion/dissemination. Since the cells can be easily recovered from the hydrogels by enzymatic digestion, they can be predifferentiated in vitro and can be used for implantation with any kind of carrier material, or the cells can be injected directly without in vitro manipulation within a gelatin matrix. It is still unclear whether the use of in vitro differentiated MSCs or the use of undifferentiated cells is more promising. Recently, Le Maitre et al. reported that MSCs from older individuals differentiate spontaneously into chondrocyte-like nucleus pulposus cells upon insertion into bovine nucleus pulposus tissue in vitro, and thus may not require additional stimulation to induce differentiation (23). If these results can be confirmed in in vivo studies, the application of MSCs will be facilitated. Our study does not provide new insights regarding this aspect, but the gelatin hydrogels were found to be a suitable material for either strategy, in vitro differentiation or in vivo injection of differentiated or undifferentiated cells.
Cell–cell contacts did not seem to be obligatory for chondrogenic differentiation in gelatin hydrogels because the cells are distributed sporadically within the gel at the low cell concentrations used in our experiments. The formation of a precartilage condensation-like structure was found not to be necessary for the induction of chondrogenesis as originally assumed by Johnstone et al. (17). In our hydrogels we observed no signs of gel contraction. The hydrogels maintained their size and shape during the whole cultivation period. Successful chondrogenic differentiation at low cell concentrations has also been demonstrated in collagen type I hydrogels (33,54). However, Nöth et al. reported a contraction of up to 75% in the collagen gels, a process that could be disadvantageous for implantation purposes (33).
We chose a low cell concentration for differentiation due to the limited nutrient supply expected within the intervertebral disc tissue. The chance of survival of vital and metabolically active cells after implantation into the disc is expected to be much higher if cell numbers do not exceed normal cell density. Very little information about cell density is available in the literature. The cell density in the nucleus pulposus is described to be anywhere from 2 to 5 million cells/cm3, with decreasing numbers due to aging and degeneration (2,3,24,31,34). It has been reported that more than 50% of all cells in adult discs are necrotic (48). Based on our own experimental data for the recovery of cells from aged disc tissue after enzymatic digestion (data not shown), we used cell concentrations comparable to those in the native tissue to avoid the onset of cell necrosis/apoptosis.
Supplementation of the gelatin hydrogel with HA did not result in a further enhancement of the in vitro chondrogenic differentiation process. The HA concentration used here was equivalent to the concentration at which HA occurs in the healthy disc (approximately 0.3–0.4%) (43). HA is already used in the treatment of degenerative joint diseases and possesses anti-inflammatory and cell-protective properties that might also have a beneficial influence on the success of cell therapy (38).
Sox-9 gene expression was found to be upregulated early on and was not essentially changed under hypoxia, similar to that described by Hardingham et al. (12). Since Sox-9 is known as the major transcription factor for COL2A1, MIA, and aggrecan genes, this appears to be in conflict to the strong increase of COL2A1, MIA, and aggrecan gene expressions. However, Sox-9 expression is not directly correlated to the expression of functional cartilage genes. Once Sox-9 reaches its maximum, matrix genes are obviously regulated by other mechanisms, too. In fact, adult articular cartilage cells have high Sox-9 even when matrix genes are downregulated, as in tissue homeostasis or under cell division in vitro (22).
Beside the expression of markers such as collagen type II and aggrecan we also found high levels of collagen type X in differentiated MSCs. Since collagen type X is known as a marker for hypertrophic chondrocytes its role during MSC differentiation has to be elucidated in further studies. Also the expression of collagen type I, a marker for connective tissue, is enhanced in differentiated MSCs. The coexpression of collagen type I and X along other chondrocyte markers has been shown in literature previously (13,20,53). The expression of collagen type X appears even briefly before collagen type II is expressed when MSC differentiation is induced in vitro, and well before other markers of hypertrophy (36), such as alkaline phosphatase or MMP-13 are rising. The role of this regulation has yet to be identified, including the possibility that the event is an in vitro artifact (47).
In addition to the carrier matrix, hypoxia may be a key element for the differentiation of MSCs in hydrogels. In MMs, areas with low oxygen tension seem to be very probable, at least in the central part of the pellets, due to the locally high cell density. An oxygen gradient has already been shown in murine adipose-derived adult stromal cells cultured as micromasses (29). In the present experiments, as elsewhere (6,54), chondrogenesis could be improved in MMs by hypoxia, resulting in increased pellet size and an upregulation of the expression of marker genes such as COL2A1, ACAN, MIA, and COL10A1. The results of gene expression analysis were confirmed on the protein level by an increased amount of sGAG deposited in the extracellular matrix, in particular when MSC expansion was performed under reduced oxygen tension before differentiation was initiated. Analogous observations could be made with hydrogels, irrespectively of the presence or absence of HA. To summarize our findings, hypoxia in expansion cultures proved to foster MSC differentiation under hypoxic conditions. This is in accordance with findings of Xu and colleagues for murine adipose-derived adult stromal cells (52) and of Zscharnack and colleagues for ovine marrow-derived MSCs (54). Hypoxic conditions during expansion may adapt the cells to the subsequent hypoxia during differentiation, resulting in improved chondrogenesis. Our data suggest that preconditioning of MSCs to hypoxia before implantation into the disc might be advantageous for the later cellular behavior, with the underlying mechanism still to be elucidated in future work. Our results with gelatin hydrogels and MMs add to the results of several publications which showed that hypoxia promotes chondrogenic differentiation of MSCs during differentiation phase (6,19,26,30). Although the studies were carried out using different cell isolation methods, experimental setups, different growth and differentiation factors, varying oxygen tensions, etc., the conclusion that hypoxia supports chondrogenesis is common to all these approaches. It is conceivable that hypoxia affects the differentiation of MSCs in ways that may be correlated with the physiological oxygen requirements of the target organ (e.g., chondrocytes in avascular cartilage have low oxygen requirements, while the vascular bone osteoblasts require elevated O2 tensions for optimal differentiation). This hypothesis is supported by studies showing that osteogenesis is diminished by low oxygen and promoted by 20% O2 (11,28). Thus, preconditioning of MSCs to hypoxic organ conditions may support the chondrogenic process and concurrently repress unintentional differentiation into the osteogenic lineage. Our data support the idea that MSCs introduced into damaged discs may not only trigger regeneration, as shown in various animal models (5,14,40,41), but may differentiate by themselves into intervertebral disc cells.
Conclusion
Our gelatin-based hydrogels represent promising biomaterials for chondrogenic differentiation of MSCs. Differentiation of MSCs is even possible at low cell concentrations without the need for a precartilage condensing mesenchyme-like structure. Chondrogenesis is further improved by hypoxia. Low oxygen tensions during expansion phase prime MSCs for a chondrogenic differentiation. Best results are obtained when the whole culture process, encompassing both expansion and differentiation, is performed under hypoxic conditions.
Footnotes
Acknowledgments
We thank Dr. Bernhard Schewe and Dr. Peter de Zwart (BG Trauma Hospital Tübingen, Tübingen, Germany) for providing bone marrow samples and Dr. Alwin Scharstuhl for the isolation of MSCs. This study was supported from the German Ministry of Education and Research (BMBF grant 0313755). Jürgen Mollenhauer is employee of TETEC AG (Tissue Engineering Technologies AG). Christoph Gaissmaier is CEO of TETEC AG.