
Abstract
Articular cartilage is an avascular tissue composed of chondrocytes, a unique cell type responsible for abundant matrix synthesis and maintenance. When damaged, it never heals spontaneously under physiological circumstances. Therefore, the delivery of mesenchymal stem cells using hydrogel has been considered for cartilage repair. This study aims at investigating the influence of in vitro chondrogenic differentiation of human adipose tissue-derived stem cells (hATSCs) on in vivo cartilage formation when associated with a cellulose-based self-setting hydrogel (Si-HPMC). hATSCs were characterized for their proliferation, surface marker expression, and multipotency. The in vitro chondrogenic potential of hATSCs cultured within Si-HPMC in control or chondrogenic medium was evaluated by measuring COL2A1, ACAN, SOX9, and COMP expression by real-time PCR. Alcian blue and type II collagen staining were also performed. To determine whether in vitro chondrogenically differentiated hATSCs may give rise to cartilage in vivo, cells differentiated as a monolayer or in pellets were finally associated with Si-HPMC and implanted subcutaneously into nude mice. Cartilage formation was assessed histologically by alcian blue and type II collagen staining. Our data demonstrate that hATSCs exhibited proliferation and self-renewal. hATSCs also expressed typical stem cell surface markers and were able to differentiate towards the adipogenic, osteogenic, and chondrogenic lineages. Real-time PCR and histological analysis indicated that Si-HPMC enabled chondrogenic differentiation of hATSCs in inductive medium, as demonstrated by increased expression of chondrogenic markers. In addition, histological analysis of implants showed that chondrogenically differentiated hATSCs (monolayers or pellets) have the ability to form cartilaginous tissue, as indicated by the presence of sulphated glycosaminoglycans and type II collagen. This study therefore suggests that an in vitro induction of hATSCs in 2D was sufficient to obtain cartilaginous tissue formation in vivo. Si-HPMC associated with autologous hATSCs could thus be a significant tool for regenerative medicine in the context of cartilage damage.
Keywords
Introduction
Articular cartilage (AC) is a highly specialized connective tissue that covers the end of bone and forms the smooth surface on joints. It is composed of chondrocytes, a unique cell type responsible for an abundant extracellular matrix (ECM) synthesis and maintenance (13). This matrix is composed of collagens and proteoglycans, mainly type II, IX, and XI collagens and aggrecan (1,24). The collagen network gives AC its shape and strength, while proteoglycans contribute to hydrophilicity and resistance to mechanical stress.
AC is an avascular and aneural tissue (25,44) and, as a consequence, damaged or degenerative AC never heals spontaneously under normal circumstances. Therefore, damage to the articular surface is irreversible and may lead to long-term joint degeneration.
Surgical treatments such as subchondral drilling or autologous periosteum (34), perichondrium (10), and osteochondral (30) tissue transplants were initially considered in early studies. However, none of these techniques resulted in a complete regeneration of native cartilage (22,25,38). Consequently, the development of cell-based therapies for the regeneration of AC has recently been contemplated (51). Among the various cell-based therapies that have been investigated, autologous chondrocyte transplantation (ACT) (11) is currently the only one that is FDA approved. This technique, however, has significant limitations such as donor site morbidity and the de-differentiation of chondrocytes towards a fibroblastic phenotype in monolayer culture (7). Consequently, this invasive surgery ultimately leads to the formation of fibrous transitory cartilage that is biochemically and biomechanically inferior to healthy articular cartilage.
For these reasons, much research has focused on finding alternative sources of cells for cartilage repair. This research has led to the identification of autologous mesenchymal stem cells (MSCs) as candidate cells for the regeneration of tissues or organs. Their self-renewal, long-term viability, and multilineage differentiation potential make them particularly suitable. Adult MSCs were initially isolated from bone marrow (4,28,33) and characterized for their ability to proliferate in culture and to differentiate into a wide variety of cell types in response to the appropriate culture system (32). However, bone marrow sampling is an uncomfortable and painful procedure and the heterogeneity of the sample, which contains both hematopoietic and mesenchymal stem cells, reduces the number of MSCs obtained. Adipose tissue, like bone marrow, derives from the embryonic mesenchyme and contains multipotent cells (56). This tissue type has the advantage of being obtained under local anesthesia with relatively little discomfort and less donor site morbidity. It also contains MSC in larger numbers than in bone marrow (27). Human adiposederived stem cells (hATSCs) have also been shown to have multilineage potential, forming bone, cartilage, and fat (3,55). hATSCs therefore provide a promising alternative to chondrocytes and bone marrow MSCs for cartilage tissue engineering (23,46).
Tissue engineering strategies combining environmental factors and biomaterials to induce the commitment of MSCs to a chondrogenic phenotype show considerable promise as a mean of regenerating cartilage (35). Three-dimensional cultures within biomaterials are known to provide a suitable environment for the induction and maintenance of the chondrogenic phenotype (14,15,51,52). To date, various biomaterials have been evaluated for cartilage repair, including protein-based, polysaccharide-based, and synthetic matrices (25). Unfortunately, some of these have been reported to induce inflammatory responses in vivo. Within this context, we have developed a self-setting hydrogel consisting of silanized hydroxypropylmethyl cellulose (Si-HPMC) (8). Si-HPMC hydrogel has previously been shown to stimulate the chondrogenic potential of chondrocytes both in vitro in 3D culture and in vivo (47,49,50).
Given these data, we became interested in the potential breakthrough that the association of hATSCs and Si-HPMC could make in the development of biomaterial-assisted cell therapy for cartilage repair.
This study therefore aimed at investigating the influence of the in vitro chondrogenic differentiation conditions of hATSCs on in vivo cartilage formation when associated with Si-HPMC hydrogel. We first characterized MSCs isolated from human adipose tissue for their propensity to self-renew, to express cell surface markers, and to differentiate into adipogenic, osteogenic, and chondrogenic lineages. Next, we evaluated the in vitro chondrogenic potential of hATSCs cultured within Si-HPMC hydrogel. To conclude, we investigated the ability of a hybrid construct combining hATSCs and Si-HPMC hydrogel to form neocartilage in vivo. To this end, hATSCs were predifferentiated in vitro in monolayers or pellets prior to injection with Si-HPMC into subcutaneous pockets in nude mice.
Materials and Methods
Materials
Hydroxypropylmethyl cellulose (HPMC) E4M1 was purchased from Colorcon-Dow chemical (Bougival, France). Glycidoxypropyltrimethoxysilane (GPTMS) was obtained from Acros (Geel, Belgium). Cell culture plastic wares were purchased from Corning BV (Schipol-Rijk, The Netherlands). Hank's balanced sodium salt (HBSS), Dulbecco's modified Eagle medium high glucose (DMEM, 4.5 g/L), phosphate-buffered saline (PBS), penicillin/streptomycin, trypsin/EDTA (0.05%/0.53 mM), l-glutamine, and superscript III kit were obtained from Invitrogen Corporation (Paisley, UK). 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), collagenase crude type IA, red blood cell lysis buffer, trypan blue, sodium l-ascorbate, vitamin D3, insulin transferrin sodium selenite (ITS) media supplement, dexamethasone, 1-methyl-3-isobutylxanthine (IBMX), indomethacin, crystal violet, alizarin red S, alcian blue, and Oil Red O were purchased from Sigma-Aldrich (St. Louis, MO). Rosiglitazone was obtained from Cayman chemical (Ann Arbor, MI). Brilliant® SYBR® Green Master Mix was obtained from Stratagene (La Jolla, CA). PCR primers were synthesized by MWG Biotech (Ebersberg, Germany). Fetal calf serum (FCS) was purchased from Dominique Dutscher (Brumath, France). Monoclonal antibody directed against human type II collagen was purchased from MP Biomedicals (Solon, OH). β-Glycerolphosphate was obtained from Calbiochem (Darmstadt, Germany). Anti-human CD29 and CD105 fluorescein isothiocyanate-conjugated antibodies and CD44, CD49d, CD90, CD34, and CD45 phycoerythrin-conjugated antibodies were obtained from Becton Dickinson (Franklin Lake, NJ). Transforming growth factor-β1 (TGF-β1) was obtained from PeproTech Inc. (London, UK). Horseradish peroxide-conjugated streptavidin (STREPTA PER) was obtained from Dako (Glostrup, Denmark). An RNeasy micro kit was purchased from Qiagen (Courtaboeuf, France). All other chemicals were obtained from standard laboratory suppliers and were of the highest purity available.
Characterization of Human ATSC
Cell Isolation
hATSCs were isolated by collagenase digestion of lipoaspirates obtained from three different patients undergoing liposuction and who had given their informed consent. All protocols were approved by the French national ethical committee. Briefly and as previously described (20,36), lipoaspirates were washed extensively with HBSS to remove debris. Washed lipoaspirates were treated with collagenase (0.025%) in HBSS for 1 h at 37°C with gentle agitation. The collagenase treatment was inactivated by adding an equal volume of DMEM containing 1% penicillin/streptomycin, 1% l-glutamine, and 10% FCS (control medium). The digested product was then centrifuged at 250 x g for 5 min to separate adipocytes from stromal cells. The supernatant was removed and cells were resuspended in the control medium and filtered through a 70-μm nylon mesh filter. The filtrate was centrifuged and cells resuspended in red blood cell lysis buffer. The lysis reaction was stopped by adding control medium. The suspension was centrifuged and cells were finally resuspended in control medium and plated at 5 × 104 cells/cm2 in 75-cm2 culture flasks. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2 and 95% air and the culture medium was replaced 24 h after seeding to remove nonadherent cells. Thereafter, the culture medium was renewed every 2–3 days. To prevent spontaneous differentiation, hATSC primary cultures (P0) were grown to 90% confluence and then detached from the cell culture flask using trypsin/EDTA. For all subsequent experiments hATSCs were used at passage 2.
Proliferation Assay
hATSCs were plated at a density of 1 × 104 cells/cm2 in 24-well plates and maintained in control medium. Cells were detached daily with trypsin/EDTA and counted using trypan blue exclusion dye. hATSCs from P0 to P3 were monitored over a 14-day period for each passage. Results were expressed as doubling time. Cell doubling time (DT) and cell doubling number (CD) were calculated from counts (N f : final cell number, N i : initial cell number) and cell culture time (CT) according to the following 2 formulae: CD = ln(N f /N i )/ln(2), and DT = CT/CD.
Clonogenic Assay for Fibroblastoid-Like Colony Formation (CFU-F)
hATSCs were plated at a density of 10 cells/cm2 in 100-mm petri dishes and cultured in the presence of control medium. After 14 days, cells were rinsed with prewarmed PBS, fixed, and stained at room temperature with 0.5% crystal violet in methanol for 30 min and afterwards gently rinsed with PBS. Colonies were counted using a phase contrast light microscope and aggregates of >50 stained hATSCs were considered as positive for CFU-F (40). Results were expressed as the percentage of cells able to form colonies.
Flow Cytometry
hATSCs (from P0 to P2) were characterized by flow cytometry using anti-human CD29 and CD105 FITC-conjugated antibodies and anti-human CD44, CD49d, CD90, CD34, and CD45 PE-conjugated antibodies (36). The adherent hATSCs were detached using trypsin/EDTA, centrifuged for 1 min at 1,200 x g, and resuspended in PBS/BSA (1%)/sodium azide (5%). Aliquots containing 2 × 105 hATSCs were incubated with antibodies for 15 min at 4°C. The suspended hATSCs were washed and then analyzed using a FACSCalibur fluorescence-activated cell sorter (Becton Dickinson, Franklin Lake, NJ). For each sample, 10,000 events were acquired and analyzed using CELLQUEST software. Results were expressed as the percentage of positive cells compared with the isotype-matched negative control antibodies on histogram plots.
Multipotent Cell Differentiation Assay
For the in vitro adipogenic differentiation of hATSCs, cells were seeded at a density of 3 × 104 cells/cm2 in six-well plates and cultured either in the presence of control or adipogenic medium for 14 days. The adipogenic medium consisted of the control medium supplemented with 1 μM dexamethasone, 0.5 mM IBMX, 200 μM indomethacin, 10 μg/ml insulin, 10 μg/ml transferin, 10 ng/ml sodium selenite, and 5 μM rosiglitazone. hATSCs were maintained at 37°C in a humidified atmosphere (5% CO2 and 95% air). Media were changed every 2–3 days. Adipogenesis of hATSCs was assessed by staining cells with Oil Red O for the lipid droplet detection. hATSCs cultured as described above were washed with ice-cold PBS and fixed in 10% formalin successively for 5 min and 1 h with fresh formalin at room temperature. The formalin was then discarded and the wells rinsed with 60% isopropanol. Once the wells had completely dried, 0.35% Oil Red O solution in 60% isopropanol was added for 10 min. Stained cells were extensively washed with deionized water to remove any nonspecific staining. Samples were visualized using a light microscope (Zeiss Axioplan 2, Göttingen, Germany) with red staining, indicating the presence of lipid droplets.
For the in vitro osteogenic differentiation of hATSCs, cells were seeded at a density of 1 × 104 cells/cm2 in six-well plates and cultured either in the presence of control or osteogenic medium for 28 days, as previously described (36). The osteogenic medium was composed of the control medium supplemented with 10 mM β-glycerophosphate, 50 μM sodium l-ascorbate, and 10 nM vitamin D3. hATSCs were maintained at 37°C in a humidified atmosphere (5% CO2 and 95% air). Media were changed every 2–3 days. Osteogenesis was evaluated by the deposition of calcified matrix that was detected using Alizarin Red S staining as previously described (36). Briefly, hATSCs cultured as described above were washed with cold PBS and stained with 2% Alizarin Red S solution for 2 min. Stained samples were extensively washed with deionized water to remove nonspecific staining. Samples were visualized using a light microscope with red staining, indicating the presence of calcium deposits.
For the in vitro chondrogenic differentiation of hATSCs, 5 × 105 cells were placed into a 15-ml polypropylene tube containing 1 ml of control medium, as previously reported (36). They were then centrifuged for 5 min at 250 x g. The tubes were fitted with vented caps to permit gas exchange, and the cell pellets were maintained at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. After 24 h, pellets of hATSCs were divided into two experimental groups and cultured either in the presence of control (CT) or chondrogenic (CH) medium. The chondrogenic medium was composed of serum-free control medium supplemented with 1% l-glutamine, 6.25 μg/ml insulin, 6.25 μg/ml transferin, 6.25 ng/ml sodium selenite (ITS), 50 nM sodium l-ascorbate, 10−8 M dexamethasone, and 10 ng/ml TGF-β1. Culture media were changed every 2–3 days for 28 days (12).
Chondrogenesis was evaluated by the production of sulphated glycosaminoglycans (GAG). GAG production was investigated on pellets by alcian blue staining. Pellets were fixed in 10% formalin and embedded in paraffin. Paraffin sections (5 μm thick) were deparaffinized with toluene, rehydrated through a graded series of ethanol, and rinsed in distilled water. Sections were then stained with alcian blue. Samples were visualized using a light microscope (Zeiss Axioplan 2, Göttingen, Germany) (36). Alcian blue staining indicated the presence of sulphated GAG.
Three-Dimensional Chondrogenic Differentiation of hATSCs Into Si-HPMC Hydrogel
Preparation of Si-HPMC Hydrogel
As previously described, the synthesis of Si-HPMC was performed by grafting 14.24% of 3-glycidoxypropyltrimethoxysilane onto E4M1 in heterogeneous medium (9). Si-HPMC powder (3% w/v) was solubilized in 0.2 M NaOH under constant stirring for 48 h. The solution was then sterilized by steam (121°C, 20 min). To allow the formation of a reticulated hydrogel, the solution was finally mixed with 0.5 volume of 0.26 M HEPES buffer. Final product is a viscous liquid at pH 7.4, allowing cell incorporation. The mixture cell/hydrogel then reticulates in approximately 30 min as described previously (9,21).
Culture of hATSCs in Si-HPMC
As previously described hATSCs were collected and gently mixed with Si-HPMC hydrogel, prepared (49) at a density of 2 × 106 cells/ml of hydrogel. The hATSC/Si-HPMC mixture (1 ml) was seeded in 12-well plates and incubated at 37°C and 5% CO2. After a 2-h incubation, control medium was added. After 24 h, hATSC/Si-HPMC constructs were separated into two experimental groups and cultured either in the presence of control or chondrogenic medium. hATSCs were maintained at 37°C in a humidified atmosphere (5% CO2 and 95% air) for 21 days. Media were changed every 2–3 days.
Analysis of the Chondrogenic Differentiation of hATSCs
For real-time PCR analysis, total RNA was extracted from the construct samples using an RNeasy micro kit in accordance with the manufacturer's instructions. After DNase I digestion, RNA was quantified using a UV-spectrophotometer (Nanodrop ND-1000, Labtech, Palaiseau, France) and the quality was determined with the Agilent Bioanalyser 2100 system. One microgram of RNA per sample was reverse-transcribed using the superscript III kit in a total volume of 20 μl. Complementary DNA (cDNA) was amplified in a total volume of 25 μl PCR reaction mix containing 12.5 μl of Brilliant® SYBR® Green Master Mix (1x) and 30 nM of SYBR green reference dye. The sequence and concentration of each primer set are provided in Table 1. The real-time PCR was carried out in a MX3000P® real-time PCR system (Stratagene) under the following conditions: 10 min at 95°C followed by 40 cycles of 30 s at 95°C, 1 min at 60°C, and 30 s at 72°C. The efficiency and specificity of each primer set were confirmed with standard curves of cycle threshold (Ct) values versus serial dilution of total RNA and melting profile evaluation. Cycle thresholds were normalized to β-actin to control for cDNA quantification differences. Results were reported as relative expression levels.
Sequences of Primer Pairs, Gene Bank Accession Numbers Used for Real-Time RT-PCR Analysis, and Size of PCR Products
For histological and immunohistological analysis, as described above, construct samples were prepared in the same manner as pellets. Tissue sections were stained with alcian blue and immunostained for type II collagen as previously described (36). Human nasal cartilage sections were used as a positive control. As a negative control, sections were processed using identical protocols, but omitting the primary antibody. Sections were then visualized using a light microscope with immunopositive areas exhibiting brown staining.
In Vivo Chondrogenic Potential of hATSCs
To investigate whether constructs associating Si-HPMC and hATSCs enable the formation of a cartilaginous tissue in vivo, we implantated constructs into subcutaneous pockets of 1-month-old nude mice (Swiss nude mice, Charles River, L'Arbresle, France). hATSCs were cultured in monolayers (10,000 cells/cm2) (2D) or in pellets (3D) in the presence of control or chondrogenic medium for 3 weeks. At the end of the culture period, cells or pellets were histologically processed for alcian blue staining. Samples were also processed for transcript analysis by real-time PCR as described above.
For implantation into nude mice, human ATSC were cultured for 3 weeks in monolayers (10,000 cells/cm2) (2D) or in pellets (3D) in the presence of control or chondrogenic medium. Cells cultured in 2D were individualized and collected by tryspin/EDTA treatment, an additional digestion step using collagenase was needed to properly disrupt the pellets. Briefly, pellets were treated with collagenase (0.25 mg/ml) in HBSS for 30 min at 37°C under constant stirring. Collagenase was inactivated by adding an equal volume of control medium. Pellets were then disrupted through repeated pipetting. Cells were finally centrifuged at 250 x g for 5 min. Individualized hATSCs (5 × 105) were gently mixed with 250 μl of Si-HPMC hydrogel prior its reticulation and implanted subcutaneously into nude mice as previously described (48). Si-HPMC was also exclusively implanted and used as a negative control. Primary horse nasal chondrocytes (HoNC) were associated (5 × 105 HoNC/250 μl Si-HPMC) with Si-HPMC and injected subcutaneously to serve as a positive control. According to a protocol modified from (47), HoNC were isolated from the nasal cartilage of an 18-month-old adult female horse. Briefly, the nasal septum was cut into small slices and digested at 37°C with 0.05% hyaluronidase in HBSS for 10 min, then with 0.2% trypsin for 15 min and with 0.2% type II collagenase for 30 min. Finally, slices were digested overnight at 37°C in 0.03% collagenase in DMEM. The suspended HoNC were plated at a density of 1 × 104 cells/cm2 in 25-cm2 culture flasks and cultured in control medium. The cells were maintained for 21 days at 37°C in a humidified atmosphere of 5% CO2 and the culture medium was changed every 2–3 days.
The six different conditions (hATSCs cultured in 2D CT, 2D CH, 3D CT, 3D CH, HoNC, and Si-HPMC alone) were tested in triplicate and nine animals received implants (two implants per animal). Animal care was provided at the experimental therapeutic unit of the Nantes medical school in compliance with European directives for conducting animal experiments. Animals were sacrificed 5 weeks after implantation and the samples were processed histologically as described above. A double-blind randomized scoring was performed by two well-versed independent examiners on each section (n = 3 per replicate) to evaluate the nodule density, the alcian blue staining and the type II collagen immunostaining intensity (a score from 0 to 4 for each criterion with the highest score awarded reflecting the best condition).
Statistical Analysis
Each experiment was repeated at least three times with similar results. Results are expressed as mean ± SEM of triplicate determinations. The comparative studies of means were performed by using one-way ANOVA followed by a post hoc test (Tukey's honestly significant difference) with a statistical significance at p < 0.05.
Results
Cell Characterization
Firstly, we aimed at characterizing mesenchymal stem cells isolated from the stromal vascular fraction (SVF) of human adipose tissue for their ability to proliferate and to self-renew. To this end, the ability of hATSCs to proliferate in culture as well as their propensity to self-renew by forming fibroblastic colonies was evaluated at different passages (from P0 to P2). hATSCs were found to exhibit fibroblast-like morphology and were able to proliferate with a doubling time of 5.15 ± 0.79 days. Moreover, this proliferation rate was constant over time and number of passages (data not shown). In addition, 1.13 ± 0.13% of cells from the SVF were able to form colonies, as measured by CFU-F assay.
Secondly, mesenchymal stem cells are known to express typical surface markers. Therefore, the expression of cell surface markers associated with adult stem cell phenotype was investigated by flow cytometry on hATSCs from passages 0 and 2. The hATSCs strongly expressed the stem cell markers CD29, CD44, CD49d, CD90, and CD105 (Fig. 1) but were negative for the hematopoietic and macrophage/monocyte cell markers CD34 and CD45 (Fig. 1). Considered together, these data demonstrate that the SVF of human adipose tissue contains a cell population exhibiting adult stem cell features such as the ability to proliferate in culture, to self-renew, and to express typical surface markers.

Characterization of human adipose tissue-derived stem cells (hATSCs) at P2. Flow cytometric analysis of hATSC for CD29, CD44, CD49d, CD90, CD105, CD34, and CD45 expression. Cytometric analysis was performed as described in the Materials and Methods section. Results are expressed as the percentage of positive cells in the whole population (n = 3). ∗p < 0.05 compared with the isotype-matched control antibodies.
Multidifferentiation Potential
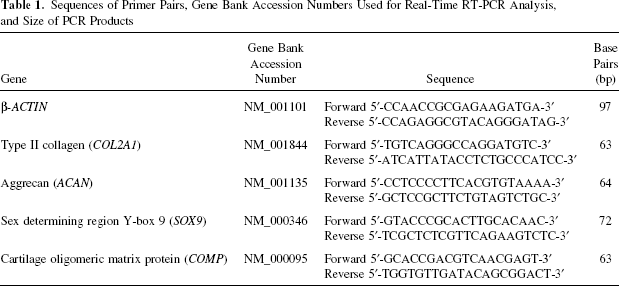
As described previously (36), another major characteristic of adult MSCs is their ability to differentiate towards multiple lineages. Adipogenesis, osteogenesis, and chondrogenesis are the three differentiation pathways most commonly described for MSCs. Consequently, to evaluate their multilineage differentiation potential, hATSCs were differentiated towards adipogenesis, osteogenesis, and chondrogenesis. Cells cultured in control medium were negative for lipid droplet detection (Fig. 2a). In contrast to this, hATSCs treated for 14 days with adipogenic medium exhibited positive Oil Red O staining, revealing the presence of vacuoles containing neutral lipids (Fig. 2b). After a 28-day period, control cultures did not form cell aggregates and were negative for mineral deposition (Fig. 2c). As expected, in the presence of osteogenic medium, hATSCs formed cell nodules positively stained by alizarin red, demonstrating the deposition of a mineralized matrix (Fig. 2d). Finally, control cells cultured in pellet did not show specific staining (Fig. 2e), whereas chondrogenically differentiated hATSCs displayed positive alcian blue staining (Fig. 2f), demonstrating a high content of sulphated GAG. Taken together, these results highlight the in vitro adipogenic, osteogenic, and chondrogenic potential of hATSCs.
Histological analysis of the multilineage differentiation capacity of hATSCs. Human ATSCs were cultured in control medium (a, c, e) or in inductive adipogenic (b), osteogenic (d), and chondrogenic (f) medium as indicated in the Materials and Methods section. Adipogenesis was detected by the formation of neutral lipid vacuoles stainable with Oil Red O (a, b). Scal bar: 50 μm. Osteogenesis was demonstrated by the deposition of a mineralized matrix indicated by alizarin red staining (c, d). Scale bar: 200 μm. Chondrogenesis was evaluated after 28 days in pellet culture by alcian blue staining (e, f). Scale bar: 200 μm.
In Vitro Chondrogenic Potential of hATSCs in Si-HPMC
Prior to testing the ability of hATSC to form cartilage in vivo when transplanted with Si-HPMC, we sought to investigate whether hATSCs could differentiate towards the chondrogenic lineage when cultured three-dimensionally within Si-HPMC hydrogel. For this purpose, hATSCs were cultured within Si-HPMC hydrogel in the presence of chondrogenic or control medium for a 28-day period. The expression of transcripts encoding COL2A1, ACAN, SOX9, and COMP was evaluated by real-time PCR. The expression levels of these mRNA substantially increased for hATSCs cultured in chondrogenic medium compared to the control condition, with a significant 8-, 123-, 4.5-, and 36-fold increase for COL2A1, ACAN, SOX9, and COMP, respectively (Fig. 3A). The production of a cartilaginous matrix was also evaluated by alcian blue staining for the presence of sulphated GAG (Fig. 3B, a, b) as well as the specific detection of type II collagen by immunostaining (Fig. 3B, c, d). The histological evaluation demonstrated that only hATSCs cultured in chondrogenic medium displayed nodules positive for sulphated GAG (Fig. 3B, b) and type II collagen (Fig. 3B, d).
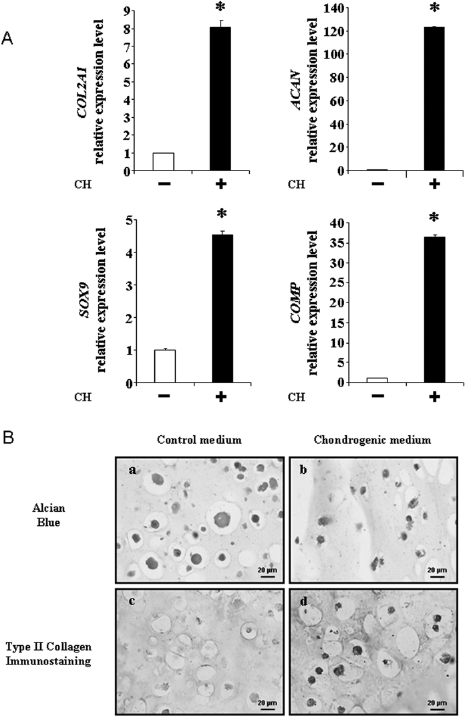
Analysis of the chondrogenic differentiation of hATSCs cultured in Si-HPMC. (A) Analysis of the expression levels of transcripts encoding for COL2A1, ACAN, SOX9, and COMP after 28 days of three-dimensional chondrogenic differentiation of hATSCs within Si-HPMC hydrogel. Expressions of the chondrogenic markers COL2A1, ACAN, SOX9, and COMP were investigated by real-time PCR, as described in the Materials and Methods section. Results are expressed as relative expression level compared to the control condition in the absence of chondrogenic medium. ∗p < 0.05. (B) Histological and immunohistological analysis of hATSCs after 21 days of three-dimensional chondrogenic differentiation within Si-HPMC hydrogel. The presence of sulphated glycosaminoglycans and type II collagen was investigated by alcian blue staining (a, b) and type II collagen immunostaining (c, d) respectively. Scale bar: 20 μm.
Taken together, these data demonstrate that the Si-HPMC hydrogel provides a three-dimensional environment allowing for the in vitro differentiation of hATSCs towards the chondrogenic lineage.
In Vivo Cartilaginous Tissue Formation
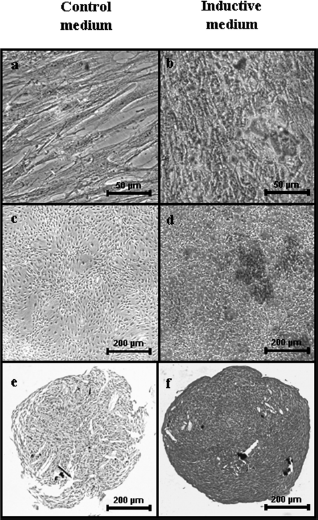
To determine whether in vitro differentially cultured hATSCs can comparably give rise to cartilaginous tissue, we subsequently conducted an in vivo implantation experiment. hATSCs were cultured for a 3-week period in monolayers (2D) or pellets (3D) and in the presence of control or chondrogenic medium. After the 3-week period of in vitro culture, chondrogenic differentiation of hATSCs was investigated. Moreover, hATSCs from the same experiment were injected in association with Si-HPMC into subcutaneous pockets of nude mice for a 5-week period.
To find out whether differentially cultured hATSCs express mRNA encoding the major phenotypic chondrocyte markers, the levels of COL2A1, ACAN, SOX9, and COMP transcripts were investigated by RT-PCR. Overall, the expression level of all the chondrogenic markers measured was the highest for cells cultured in pellets and in the presence of chondrogenic medium (Fig. 4A). In fact, the expression levels of COL2A1, ACAN, SOX9, and COMP were significantly higher in 3D chondrogenic than in 2D chondrogenic and 3D control conditions. Of note, inductive medium favored the expression of chondrogenic markers (COL2A1 and COMP) regardless of whether hATSCs were grown in monolayers or pellets. In addition, 3D culture was found to stimulate COL2A1 and SOX9 independently of the presence of control or chondrogenic medium. To further document the chondrogenic differentiation of hATSCs, we also assessed whether hATSCs produced GAG. The histological analysis revealed that hATSCs cultured in monolayers were unable to synthesize GAG even in the presence of chondrogenic medium (Fig. 4B). On the contrary, the hATSCs cultured in pellets and in the presence of chondrogenic medium expressed GAG (Fig. 4C, b). We therefore next analyzed the production of type II collagen in these conditions. The accumulation of this typical cartilage matrix protein was shown for the 3D-cultured and chondrogenically differentiated hATSC (Fig. 4C, d). These results indicate that only 3D pellet culture in the presence of chondrogenic medium supports the concomitant production of GAG and type II collagen in the ECM.

Analysis of 2D and 3D differentiation of hATSCs. (A) Analysis of the expression levels of transcripts coding for COL2A1, ACAN, SOX9, and COMP in hATSCs after 21 days of chondrogenic differentiation in monolayer or three-dimensional pellet culture. Expression of the chondrogenic markers COL2A1, ACAN, SOX9, and COMP was investigated by real-time PCR. Results are expressed as relative expression level compared to the control condition in the absence of chondrogenic medium. ∗p < 0.05 compared to control medium (-) for the same dimension and #p < 0.05 compared to 2D dimension for the same medium. (B) Histological and immunohistological analysis of hATSC after 21 days of chondrogenic differentiation in monolayer culture. The presence of sulphated glycosaminoglycans was investigated by alcian blue staining. Scale bar: 200 μm. (C) Histological and immunohistological analysis of hATSC after 21 days of chondrogenic differentiation in three-dimensional pellet culture. The presence of sulphated glycosaminoglycans and type II collagen was investigated by alcian blue staining (a, b) and type II collagen immunostaining (c, d), respectively. Scale bar: 200 μm.
Ultimately, in order to ascertain whether hATSCs from the same experiment were able to form cartilaginous tissue in vivo, they were injected in association with Si-HPMC into the subcutaneous pockets of nude mice. After 5 weeks of implantation, the mice were sacrificed and explant sections were stained with alcian blue as well as immunostained for type II collagen. The histological examination revealed the formation of nodules for hATSCs that had been cultured in the presence of chondrogenic medium as either monolayers (2D) (Fig. 5A, c, d) or pellets (3D) (Fig. 5A, g, h). These nodules exhibited positive alcian blue staining and type II collagen immunostaining, suggesting the production of a cartilaginous matrix. On the contrary, in the presence of control medium (Fig. 5A, a, b, e, f), no nodule formation could be detected. Primary horse nasal chondrocytes used as positive controls were also implanted and displayed chondrocyte nodule formation containing sulphated GAG and type II collagen (Fig. 5A, I, j). Si-HPMC used exclusively as a negative control did not show tissue formation (data not shown). To quantitatively assess the formation of nodules and the production of sulphated GAG and type II collagen, a histological scoring system was elaborated. Such scoring confirmed a significant difference between hATSC cultured in monolayers or in pellets and in chondrogenic medium in comparison to their respective controls (Fig. 5B). Of note, while the histological features of the nodules were different, no statistical difference was observed between the 2D and 3D groups for the same culture medium (Fig. 5B). These results indicate that hATSCs cultured in chondrogenic medium, regardless of its dimension, and associated with Si-HPMC hydrogel, are able to form nodules with cartilaginous features, in a subcutaneous site.

Cartilaginous tissue formation of hATSCs after subcutaneous implantation into nude mice. (A) Histological and immunohistological analysis of hATSC/Si-HPMC subcutaneous implants. hATSC cultured either in monolayers (2D) or in pellets (3D) in the presence of control (CT) or chondrogenic (CH) medium for 21 days were associated with Si-HPMC and implanted subcutaneously into nude mice for 5 weeks. Horse nasal chondrocytes (HoNC) associated with Si-HPMC were used as a positive control. Histological sections were stained with alcian blue (a, c, e, g, i) and immunostained for type II collagen (b, d, f, h, j). Scale bar: 50 μm. (B) Histological scoring of hATSC/Si-HPMC subcutaneous implants. Histological sections were evaluated for nodule density (black), alcian blue staining intensity (white), and type II collagen immunostaining intensity (striped). Three sections of each condition were randomly and double-blindly scored (score from 0 to 4 for each criterion: 0, absence; 1, low; 2, intermediate; 3, marked; 4, intense). ∗p < 0.05 compared with the respective control medium. #p < 0.05 compared with the respective 2D condition.
Discussion
When damaged, AC always fails to heal spontaneously under physiological circumstances. Therefore, injuries are irreversible and may lead to cartilage degeneration and to loss of joint function. Within this context, strategies associating autologous MSC such as hATSCs and biomaterials show considerable promise for repairing articular cartilage.
In the present study, our objectives were to investigate whether a Si-HPMC hydrogel could be a suitable scaffold for (i) supporting hATSC chondrogenic differentiation in vitro and (ii) allowing the transplantation of differentially precommitted cells and the formation of a cartilaginous tissue in nude mice.
In a first set of experiments, we sought to characterize adherent cells from the stromal vascular fraction of human adipose tissue. Proof of their proliferation, self-renewal, expression of typical MSC surface markers as well as their capacity for multilineage differentiation was obtained by our study. These results indicate that hATSCs display several stem cell characteristics (17). Our subsequent objective was to associate hATSCs with Si-HPMC hydrogel for cartilage tissue engineering. To this end, the question was firstly posed as to whether Si-HPMC may be a suitable scaffold for supporting the chondrogenic differentiation of hATSCs. When placed in chondrogenic conditions, hATSCs cultured within Si-HPMC hydrogel were still able to undergo chondrogenic differentiation. The major chondrocyte phenotypic markers, COL2A1, ACAN, SOX9, and COMP, were found to be expressed at the mRNA level. Moreover, sulphated GAGs and type II collagen were accumulated within the extracellular matrix. These data demonstrate that Si-HPMC hydrogel is a biomaterial providing a 3D environment able to support the in vitro chondrogenic differentiation of hATSCs. This beneficial effect of 3D culture within Si-HPMC hydrogel is in all likelihood due to its ability to allow rounded cell morphology, one of the characteristics critical to obtaining the chondrocytic phenotype (2,19). Additionally, it is well known that a 3D environment involving cell/cell or cell/matrix interactions is required for proper chondrogenic differentiation (7,19,29,31). Moreover, 3D culture within matrix can also be a more potent differentiation cue for MSCs than standard induction cocktails, as has been previously demonstrated for osteogenesis and adipogenesis (5,6,16).
The ideal biomaterial for stem cell-based cartilage tissue engineering should not only allow the maintenance of the chondrocyte differentiation state or the chondrogenic differentiation of MSCs but should also enable their transplantation via a mini-invasive surgical protocol in vivo.
Consequently, to decipher whether Si-HPMC hydrogel could represent this ideal scaffold, we investigated its ability to support the transfer of hATSC and the formation of cartilage tissue in vivo. In a first attempt, among the various in vitro conditions tested (2D CT, 2D CH, 3D CT, and 3D CH), the question was to know which one might meet the efficient criteria to form cartilage tissue and its feasibility within the context of a cartilage tissue engineering application. Based on our real-time RT-PCR and histological analyses, hATSCs cultured in 3D CH were considered the most suitably committed cells as compared to those cultured in 2D or in 3D but in the absence of chondrogenic medium. These results are in accordance with the numerous studies using pellet culture systems to examine the in vitro chondrogenesis of MSC (41,42).
Interestingly, after 5 weeks in vivo, hATSCs cultured in chondrogenic medium, either in 2D or 3D, exhibited positive staining for sulphated GAG and type II collagen and were comparably scored. In contrast, noninduced hATSCs (without chondrogenic medium), grown in monolayers or 3D pellets, failed to exhibit sulphated GAG and type II collagen production. Consistently, a 3D matrix alone appears insufficient to initiate the chondrogenic differentiation of hATSC in the absence of a specific induction medium. On the contrary, cartilaginous tissue formation was achieved with hATSCs treated with a chondrogenic medium in both 2D monolayer and 3D pellet culture. Furthermore, while hATSCs cultured in 2D chondrogenic medium failed to achieve complete chondrogenic differentiation in vitro, once in vivo within Si-HPMC they gave rise to a cartilaginous tissue to the same extent as hATSCs predifferentiated in 3D. The discrepancy of nodule appearance between 2D CH, 3D CH, and HoNC, however, requires careful analysis. It seems reasonable to speculate that as a function of their origins and commitment stages, cells do not probably respond to 3D environment and biomaterial properties (stiffness, elasticity?) in the same manner (18). This intriguing point should be paid further attention. The 3D pellet culture system is widely used to induce chondrogenic differentiation in vitro (26) albeit that a number of serious drawbacks have to be surmounted so that 3D pellet culture can be considered for clinical application. Firstly, this MSC pellet culture system mimics the endochondral ossification process. It has thus been demonstrated that the in vitro chondrogenesis of MSCs is characterized by the induction of a broad panel of hyaline cartilage molecules concomitant with early onset of hypertrophic markers such as COL10A1 and alkaline phosphatase (ALP), markers for terminal differentiation (MMP-13), and osteoblasts (osteopontin and bone sialoprotein II) (37,43,53,54). This phenomenon could ultimately cause extensive in vivo calcification of the ECM, vascular invasion, and thus instability of the ectopic transplants (39). However, the long-term phenotypic stability along with the functional suitability in vivo and the adoption of a nonhypertrophic chondrocyte phenotype are paramount for an effective and sustained cartilage repair. Secondly, it is necessary to individualize the cells by disrupting the pellets in order to associate them evenly in the biomaterial. This is a limiting step for clinical application.
The rather similar histological scoring obtained for 3D- and 2D-induced hATSCs suggests that chondrogenic medium could initiate the chondrogenic commitment of hATSCs, even in monolayer culture. This discrepancy observed between the in vitro and in vivo results may arise from different commitment stages in vitro, which were waived after 5 weeks in a favorable in vivo environment within Si-HPMC. It seems therefore reasonable to assume that a simple commitment of hATSCs towards the chondrogenic phenotype in vitro would be sufficient to enable cells to undergo chondrogenic differentiation, once placed in favorable conditions in vivo. The subcutaneous site is however rather far from a cartilage repair situation due to the lack of mechanical constraints that are known to favor chondrogenic commitment of MSCs (45). Therefore, further studies must be performed and should address the propensity of hATSCs, induced chondrogenically or not, to repair a load-bearing AC defect when implanted with Si-HPMC.
In summary, our study demonstrates that hATSCs are able to differentiate towards the chondrogenic lineage in vitro when cultured in a 3D environment in pellets or within a polysaccharide hydrogel, and to form cartilaginous tissue in vivo. Moreover, this study suggests that an in vitro 2D commitment of hATSCs would be sufficient to obtain cartilaginous tissue formation in vivo, which could provide an easier use of hATSCs for cartilage tissue engineering strategies. The long-term objective of this study is to develop a clinically relevant cartilage tissue engineering procedure using hATSCs as a source of autologous chondrogenic cells and Si-HPMC as a vehicle able to maintain these cells and foster chondrogenesis at the damaged site. Further experiments in suitable animal cartilage lesion models therefore require further attention.
Footnotes
Acknowledgments
This study was financed by grants from the “foundation Arthritis Courtin,” the “Société Française de Rhumatologie,” “ANR young researchers, project scartifold,” ANR Tecsan “Chondrograft,” the “fondation de l'avenir pour la recherche médicale appliquéée,” and the INSERM U791. C.M. and S.P. received a fellowship from “Région Pays de la Loire, program Bioregos I and Bioregos II.” The authors also gratefully acknowledge Dr. F. Lejeune (clinique Brétéché Nantes), K. Rouger (INRA UMR 703-ENVN), R. Josien, M. Heslan, S. Rémy, I. Anegon (INSERM U643) for their helpful assistance in flow cytometry experiments, and M. Gatius-Perré for helpful discussions. Authors also thank J. Ashton-Chess for critical reading of the manuscript. Dr. C. Vinatier is an employee of GRAFTYS SA company. Dr. J. Guicheux owns shares and is a consultant to the GRAFTYS SA company.