
Abstract
Endothelial progenitor cells (EPCs) consist of two different subpopulations named early (eEPCs) and late EPCs (lEPCs) that are derived from CD14+ and CD14- circulating cells, respectively. These cells are regularly cultured over fibronectin-coated surfaces in endothelial basal medium (EBM)-2 supplemented with insulin-like growth factor (IGF-1), vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and fibroblast growth factor (FGF). We have developed a new and simplified method for culturing human EPCs obtained from peripheral blood and tested their ability to preserve cardiac function following infarction. We first demonstrated that eEPCs derived from human peripheral blood mononuclear cells (PBMCs) and cultured in EBM-2 medium supplemented with autologous serum (10%) over fibronectin-coated surfaces (10 μg/ml) in the presence of IGF-1 (50 ng/ml) only, have a secretome similar to eEPCs cultured under regular conditions with IGF-1, VEGF, EGF, and FGF. Our data also indicate that IGF-1 modulates PBMC secretome in a dose-dependent manner. In another series of experiments, we showed that PBMCs cultured in suspension in bags (S-PBMCs) in basal medium supplemented with fibronectin and IGF-1 secrete significant amounts of stem cell factor (SCF, 31.3 ± 3.1 pg/ml)), hepatocyte growth factor (HGF, 438.6 ± 41.4 pg/ml), soluble tumor necrosis factor receptor 1 (sTNFR1, 127.1 ± 9.9 pg/ml), VEGF (139.3 ± 9.6 pg/ml), and IGF-1 (147.2 ± 46.1 pg/ml) but very low levels of TNF-α (13.4 ± 2.5 pg/ml). S-PBMCs injected intravenously into NOD SCID mice migrated to the injured myocardium, reduced cardiac fibrosis, enhanced angiogenesis, and preserved cardiac function after myocardial infarction (MI) in a manner similar to eEPCs cultured under standard conditions. In conclusion, we show in this study a refined and optimized method for culturing eEPCs. Our data indicate that S-PBMCs are composed of several cell populations including eEPCs and that they secrete high amounts of antiapoptotic, anti-inflammatory, and proangiogenic factors capable of preserving cardiac function following MI.
Keywords
Introduction
Endothelial progenitor cells (EPCs) were first identified by Asahara et al. as a subset of CD34+ hematopoietic progenitor cells (3). EPCs are isolated from circulating mononuclear cells (3,25), bone marrow (27), and cord blood (24). They express antigens such as CD14, CD34, vascular endothelial growth factor receptor 2 (VEGF-R2) and CD133 shared by hematopoietic stem cells together with endothelial cells and exhibit numerous endothelial properties (9,12,21). Controversy exists with respect to the identification and the origin of endothelial progenitor cells. Overall, there is consensus that EPCs consist of two different subpopulations with different morphologies and growth patterns, termed early and late EPCs. Early EPCs (eEPCs) are derived from CD14+ subpopulations of peripheral blood mononuclear cells (PBMCs) and exhibit a spindle-like morphology when cultured during 1 week under angiogenic conditions. eEPCs virtually do not proliferate and are not able to form vascular structures in vitro (12,26,35,39). Late EPCs (lEPCs) are derived from CD14 fraction. They exhibit cobblestone morphology after 3–4 weeks of culture under angiogenic conditions. lEPCs form vascular networks in vitro and in vivo (12,39).
EPCs have been investigated as a new strategy in regenerative medicine for ischemic diseases. Several clinical trials have been performed following the promising results obtained in animal models of ischemia-induced cardiovascular injury (8,15,16). In one study, autologous AC133+ bone marrow cells were injected into tissue adjacent to the ischemic area during coronary artery bypass surgery in six patients who had suffered a myocardial infarction (MI). The procedure enhanced the global left ventricular function and improved infarcted tissue perfusion (31). Transplantation Of Progenitor Cells And Regeneration Enhancement in Acute Myocardial Infarction (TOPCARE-AMI) investigators reported greater left ventricular ejection fraction (LVEF) and viability following intracoronary injection of bone marrow-derived cells and circulating progenitor cells in a group of patients (29). Interestingly, there was no difference between groups in the TOPCARE-AMI study in which patients were treated with bone marrow-derived cells or circulating progenitor cells, suggesting that both cell populations may be equipotent in their ability to improve myocardial function after an MI. In the BOne marrOw transfer to enhance ST-elevation infarct regeneration (BOOST) trial, accelerated LVEF recovery after an MI was found following bone marrow cell injection and coronary intervention (23).
eEPCs are typically cultured for 1 week in fibronectin-coated surfaces in endothelial basal medium (EBM)-2 supplemented with VEGF, insulin like growth factor (IGF-1), epidermal growth factor (EGF), and fibroblast growth factor (FGF) (2,5,14,17,19,30,33,37). eEPC-adherent culture production protocols are cumbersome and typically require specialized infrastructure and personnel, limiting widespread use. In an effort to simplify eEPC production while retaining therapeutic potency, we refined the method of eEPC culture. Here, we show that PBMCs cultured in suspension in bags (S-PBMCs) in EBM-2 supplemented only with fibronectin and IGF-1, secrete antiapoptotic, anti-inflammatory, and proangiogenic factors that we show have a positive impact on cardiac function and remodeling following MI.
Materials and Methods
Reagents
ELISAs for tumor necrosis factor (TNF)-a, stem cell factor (SCF), VEGF, soluble TNF receptor (sTNF-R)-1, IGF-1, and hepatocyte growth factor (HGF) as well as anti-CD4, anti-CD8, anti-CD14, anti-CD34, and anti-CD56 antibodies used for fluorescence activated cell sorting (FACS) were obtained from R&D Systems (Minneapolis, MI, USA). EBM-2, human (h)EGF, human FGF, VEGF, and IGF-1 were from Lonza (Walkersville, MD, USA). ELISA for cardiac troponin I (cTNI) was from Life Diagnostics (Westchester, PA, USA). RPMI was purchased from Hyclone (Logan, UT, USA). Permalife cell culture bags were from Origen (Helsingborg, Sweden).
Cell Isolation and Culture
Human PBMCs were obtained by Ficoll gradient separation of healthy donor apheresis. The continuous flow cell separator machine COBE Spectra Version 6.1 (COBE Laboratories, Lakewood, CO, USA) was used to collect white blood cells from peripheral blood as previously reported. The machine was set up and primed according to the manufacturer's instructions. Bilateral peripheral venous access via a basilic or cephalic vein was used to perform the collection and low molecular weight heparin was used as the anticoagulant. This completely automated procedure is based on parameters entered at the start of the procedure (hematocrit, weight, and height). Usually, two times total blood volume is processed over approximately 3–4 h at a rate of less than 65 ml/min. No prior mobilization regimens were given. The cells were harvested into the collection bag and used for study purposes. The study was approved by the Royal Victoria Hospital ethics committee (McGill University Heath Centre) and a signed informed consent was obtained from each participant. PBMCs were either seeded in T75 flasks (regular adherence condition) or cultured in suspension in 30-ml bags. Medium used to culture eEPCs consisted of EBM-2 supplemented with 10% autologous human serum, 1% Pen/Strep, 0.04% hydrocortisone, 0.1% heparin, and 0.1% ascorbic acid in presence of IGF-1 (50 ng/ml unless otherwise indicated), EGF (10 ng/ml), FGF (50 ng/ml), VEGF (50 ng/ml), and fibronectin (10 μg/ml unless otherwise indicated). Monocytes were isolated using the EasySep human monocyte purification kit (Stemcell Technologies, Vancouver, BC, Canada) and cultured under adherence conditions in RPMI-based medium supplemented with 10% donor serum, 1% HEPES, 1% sodium pyruvate, and 1% Pen/Strep. For cells cultured under adherence conditions (eEPCs and monocytes), medium was changed at day 3 following cell separation and was then changed each day. For cells cultured in suspension (S-PBMCs) no medium change was performed. Cells were kept in culture for 3 days (S-PBMCs) or 1 week (eEPCs and monocytes) at 37°C in a tissue culture incubator (5% CO2). Human serum was prepared by incubating donor plasma supplemented with CaCl2 (20 mM) at 37°C under 5% CO2 for 3–4 h.
Enzyme-Linked Immunosorbent Assay (ELISA)
ELISA was performed on eEPC-, S-PBMC-, and monocyte-conditioned media following the supplier protocol. Data are normalized to the number of CD14+ cells in each population and are reported in pg/ml/106 cells/24 h.
Cell Labeling for FACS Analysis
Cells were resuspended in PBS containing 1% fetal bovine serum (FBS) buffer and incubated with phycoerythrin (PE) - or fluorescein isothiocyanate (FITC)-conjugated antibodies for 2 h at 4°C. FACS was performed using a FACScalibur analyzer (BD Bioscience).
Animal Models
All experiments were carried out in agreement with local guidelines for the care and use of laboratory animals and in accordance with the guidelines of the McGill University animal care authority. Investigations conform to the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. In vivo experiments were performed in eight NOD.CB17-Prkdcscid [also known as nonobese diabetic severe combined immunodeficient (NOD SCID)] mice per group. MI was induced by ligation of left anterior descending coronary artery as previously described. Previous publications demonstrated that injection of purified monocyte derivatives have major regenerative properties by secreting growth factors, cytokines, and metalloproteinases (MMPs) (5,11,28). Based on the FACS analysis for the CD14 marker, PKH26-labeled eEPCs and S-PBMCs were resuspended in PBS for injection respectively at a concentration of 3 × 104 and 105 cells/μl of PBS to compare equicellular amounts of CD14+ cells in both cell preparations. A total of 6 × 105 eEPCs or 2 × 106 S-PBMCs, or the equivalent PBS volume, were injected 6 days after MI per mouse through the jugular vein with a nontraumatic 30- and 27-gauge needle, respectively (Hamilton, USA).
Criteria of Inclusion and Exclusion in the MI Study
Results presented in this study include mice with the same heart rate and similar infarct sizes. The infarct size was estimated by determination by ELISA of cardiac troponin I (cTnI) in mouse plasma collected 24 post-MI as previously described (7,22). Analysis of echocardiograms and study of myocardial histology were performed on mice with heart rates of 564 ± 18.6 bpm and a cTnI concentration of 30.2 ± 2.2 ng/ml.
Echocardiography
Echocardiography was performed as described previously with some modifications, 1 month after the induction of MI. Two-dimensional guided M-mode echocardiography was performed under light isoflurane anesthesia (0.75–1% isoflurane, 1 L/min O2) using a Visual Sonic VEVO 770 ultrasound machine and a RMV™ 707B High Frame Rate Scanhead with a center frequency of 30 MHz. The percentages of isoflurane and O2 flow were adjusted to obtain the fastest heart rate (HR) possible. Left ventricle (LV) end-diastolic and end-systolic internal diameter (LVIDd and LVIDs), end-diastolic interventricular and LV posterior wall thickness (IVSd and LVPWd), and fractional shortening (FS) were determined as described previously (1,13).
Heart Dissection and Histological Analysis of EPC Migration
Mice were anesthetized with isofurane and a cut was made in the peritoneum to localize the posterior vena cava (PVC). Eight hundred microliters of 0.1 M KCl was injected through the PVC to stop the heart in diastole. An incision was performed over the left thoracic area. Muscles over the ribs were delicately separated to visualize the heart. Pericardium was carefully opened and the heart was removed. Harvested hearts were then injected with 0.5 ml of PBS/1% heparin via the superior vena cava. For the evaluation of the migration of eEPCs or S-PBMCs to the injured myocardium, hearts were mounted in OCT and sectioned at the histology facility of Immunology and Cancer Research Institute of the University of Montreal (QC, Canada). Frozen sections were then labeled with DAPI and observed using a Leica DM-LB2 microscope.
Statistical Analysis
Data are presented as means ± SEM. Differences among groups were statistically analyzed using analysis of variance (ANOVA). Tests were followed up by Bonferroni correction within groups. Statistical analyses were performed using GraphPad software. A value of p < 0.05 was considered significant.
Results
IGF-1 Is Required to Induce eEPC Differentiation
eEPCs are commonly cultured in EBM-2 medium supplemented with IGF-1, VEGF, FGF, and EGF. In a first experiment, we evaluated whether all these growth factors were required to induce eEPC differentiation. eEPCs derived from peripheral blood share several markers with myelomonocytic cells but have different secretome profiles (6,27). Therefore, we cultured PBMCs for 1 week in fibronectin-coated flasks with either IGF-1, VEGF, FGF, or EGF and analyzed their secretome relative to eEPCs cultured in presence of all four growth factors and to monocytes. Our results show that IGF-1 is capable alone of inducing PBMCs to secrete significant amounts of growth factors and cytokines (Fig. 1). PBMCs (106) cultured with IGF-1 secreted, in 24 h, 34.5 ± 4.2 pg/ml of SCF, 192.3 ± 17.3 pg/ml of VEGF, 143.2 ± 11.6 pg/ml of sTNFR1, 166.2 ± 7.9 pg/ml of IGF-1, and 1131 ± 276 pg/ml of HGF. Treatment of PBMCs with IGF-1 inhibited TNF-α secretion. The levels of growth factors and cytokines secreted by PBMCs cultured only with IGF-1 were comparable to those produced by eEPCs cultured with all four growth factors. PBMCs cultured with VEGF, FGF, or EGF secreted low levels of growth factors and cytokines in comparison with eEPCs and PBMCs cultured with IGF-1.

Secretome of adherent PBMCs cultured for 1 week under angiogenic conditions. (A) Peripheral blood mononuclear cells (PBMCs) were isolated from apheresis obtained from healthy donors using Ficoll gradient. Some cells were cultured either with insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), or epidermal growth factor (EGF). Some cells were cultured with all the above-mentioned growth factors [early endothelial progenitor cell (eEPC) group]. Purified monocytes (Mono) were cultured without any growth factor and were used as control. (B–G) Concentration of tumor necrosis factor-α (TNF-α), stem cell factor (SCF), VEGF, soluble TNF receptor 1 (sTNFR1), IGF-1, and hepatocyte growth factor (HGF) in the conditioned medium of each cell preparation reported in pg/ml/106 cells/24 h. Results are normalized to the concentration of cytokines and growth factors in the cell-free medium. Data are reported as mean ± SEM (n = 3). *,#Statistically significant difference between groups with the same symbol: p < 0.05.
IGF-1 Modulates the eEPC Secretome
The aim of the study is to test a new and simplified method for culturing human eEPCs obtained from peripheral blood. We identified the optimal concentration of IGF-1 to induce secretion of SCF, VEGF, sTNFR1, IGF-1, and HGF. Basically, PBMCs were seeded in fibronectin-coated flasks in EBM-2 supplemented with different concentrations of IGF-1. Data presented in Figure 2 indicate that IGF-1 modulates the PBMC secretome in a dose-dependent manner. eEPCs are typically cultured with 50 ng/ml of IGF-1 among other growth factors. We thus evaluated whether lower doses of IGF-1 (25, 12.5, and 6.25 ng/ml) would affect eEPC secretome. Figure 2 indicates that concentration of IGF-1 normally used to culture eEPCs (50 ng/ml) corresponds to the lowest dose to induce the secretion of growth, antiapoptotic, and proangiogenic factors (optimal dose).

Effect of IGF-1 concentration on the PBMC secretome. Concentrations of TNF-α (A), SCF (B), VEGF (C), sTNFR1 (D), IGF-1 (E), and HGF (F) in the conditioned medium of PBMCs cultured 1 week under adherence conditions on fibronectin-coated flasks in endothelial basal medium (EBM-2) supplemented with various concentrations of IGF-1. Data are expressed in pg/ml/106 cells/24 h. Results are normalized to the concentration of cytokines and growth factors in the cell-free medium. Data are reported as mean ± SEM (n = 3). *Statistically significant difference between groups with this symbol: p < 0.05.
Fibronectin Modulates the eEPC Secretomes
In order to verify whether fibronectin was required to induce eEPC differentiation, PBMCs were seeded in flasks coated with fibronectin at different concentrations and cultured in EBM-2 supplemented with 50 ng/ml IGF-1. Our data indicate that fibronectin modulates the PBMC secretomes and that the optimal concentration of fibronectin was 10 μg/ml (Fig. 3).
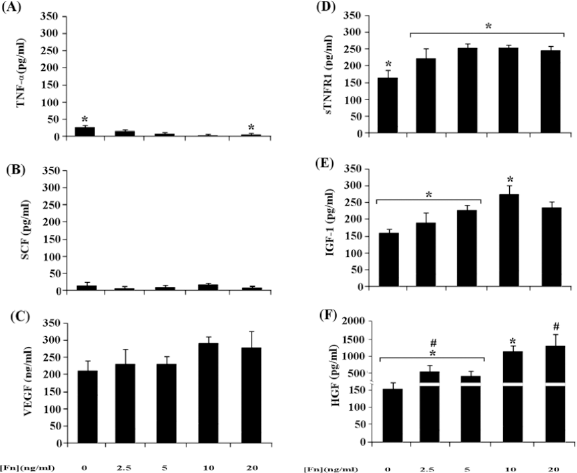
Effect of fibronectin concentration on the PBMC secretome. Concentrations of TNF-α (A), SCF (B), VEGF (C), sTNFR1 (D), IGF-1 (E), and HGF (F) in the conditioned medium of PBMCs cultured 1 week under adherence conditions in EBM-2 supplemented only with IGF-1 in flasks coated with fibronectin at various concentrations. Data are normalized to the concentration of cytokines and growth factors in the cell-free medium. Results are expressed in pg/ml/106 cells/24 h and are reported as mean ± SEM (n = 3). *,#Statistically significant difference between groups with the same symbol: p < 0.05.
S-PBMCs Secrete Anti-inflammatory, Antiapoptotic, and Proangiogenic Factors
In another series of experiments we investigated the secretome of PBMCs cultured in suspension with 50 ng/ml IGF-1 and 10 μg/ml fibronectin in bags (S-PBMCs) compared to eEPCs cultured under regular conditions, and monocytes (Fig. 4A). We compared equicellular amounts of CD14+ cells in eEPC and S-PBMC preparations. Our results indicate that S-PBMCs secreted, in 24 h, 31.3 ± 3.1 pg/ml of SCF, 139.3 ± 9.6 pg/ml of VEGF, 127.1 ± 9.9 pg/ml of sTNFR1, 147.2 ± 46.1 pg/ml of IGF-1, and 438.6 ± 41.4 pg/ml of HGF. The levels of growth factors and cytokines secreted normalized to the number of CD14+ cell contained in the S-PBMC population were comparable to those produced by eEPCs except for VEGF and HGF. Indeed, eEPCs secreted 1.6-and 3-fold more VEGF and HGF, respectively, than S-PBMCs.

Secretome of PBMCs cultured for 3 days in suspension. (A) PBMCs were isolated from apheresis obtained from healthy donors using a Ficoll gradient. Some cells were cultured in suspension with IGF-1 and fibronectin only (S-PBMCs). Some cells were cultured with IGF-1, VEGF, FGF, and EGF (EPC group). Purified monocytes (Mono) were cultured without any growth factor and were used as control group. Concentration normalized to the number of CD14+ cells are reported in pg/ml/106 cells/24 h of TNF-α, SCF, VEGF, sTNFR1, IGF-1, and HGF in the conditioned medium of S-PBMCs, eEPCs, and monocytes (Mono) in (B–G). Data are normalized to the concentration of cytokines and growth factors in the cell-free medium. Results are reported as mean ± SEM (n = 3). *Statistically significant difference between groups with this symbol: p < 0.05.
Intravenous Injection of S-PBMCs Preserved Cardiac Function After MI
We first characterized the lymphomyeloid cell population in eEPCs and S-PBMCs. eEPCs were composed of 93% CD14+ and 21% CD34+ cells whereas CD4+, CD8+, and CD56+ cells were virtually absent. S-PBMCs were composed of 58% CD4+, 39% CD8+, 32% CD14+, 22.4% CD34+, and 29% CD56+ cells (Fig. 5A). Based on FACS analysis for the CD14 marker, we treated NOD SCID mice with PKH26-labeled eEPCs and S-PBMCs, respectively, at a concentration of 3 × 104 and 105 cells/μl of PBS to compare equicellular amounts of CD14+ cells in both cell preparations and evaluated the cardiac outcome. Figure 5B and C shows that injected cells migrated to the injured myocardium. There were threefold (p = 0.01) more S-PBMCs than eEPCs per heart section, consistent with the amount of injected cells. Injection of S-PBMCs reduced cardiac fibrosis 2.8-fold (p = 0.02) (Fig. 5D) and enhanced the number of capillaries in the border and scar zones 1.9-fold (p = 0.03) and 1.6-fold (p = 0.03), respectively, in comparison to the MI group (Fig. 5E, H). Left ventricle fractional shortening (% LVFS) of NOD SCID mice injected with S-PBMCs and eEPCs improved 44.2% (p = 0.001) and 43.3% (p = 0.001), respectively, relative to the untreated MI group (Fig. 5F, G).

Effect of the injection of S-PBMCs on cardiac function following myocardial infarction (MI). (A) FACS analysis for CD4, CD8, CD14, CD34, and CD56 lymphomyeloid cells present in S-PBMCs and eEPCs. (B) Number of S-PBMCs or eEPCs injected intravenously 6 days following MI/mm2 of nonobese diabetic severe combined immunodeficient (NOD SCID) mouse heart sections. (C) Representative photos of area of heart cross sections of mice injected with S-PBMCs or eEPCs (PKH26, red labeling). Nuclei are stained in blue (DAPI). (D) Sirius red-stained surface as percentage of heart section area. (E) Capillary density (number/mm2) within the scar and the border zones. (F) Left ventricle fractional shortening (%LVFS) of NOD SCID mice injected with S-PBMCs or eEPCs. (G) Size in millimeters of left ventricle internal dimension in systole (LVIDs), left ventricle internal dimension in diastole (LVIDd), interventricular septum in systole (IVSs), and left ventricle posterior wall in systole (LVPWs) of NOD SCID mice injected with S-PBMCs or EPCs. (H) Representative cross sections of hearts stained for hematoxilin & eosin (H&E), Sirius red (SR), and CD31. Experiments were performed in NOD SCID mice after sham treatment, MI and S-PBMC or eEPC injection. nd: not detected. Results are reported as mean ± SEM (n = 8). *,#, Statistically significant difference between groups with the same symbol: p < 0.05.
Discussion
Neovascularization is a major challenge in tissue regeneration research. EPCs have been suggested to play an important role in postnatal angiogenesis and arteriogenesis (6). Early reports described the isolation and culture of eEPCs from mononuclear cells. These cells possessed endothelial cell morphology and expressed hematopoietic and endothelial markers after 3–7 days in culture (32). The regular procedure for preparation of eEPCs includes the use of adherence culture of PBMCs or applies immunomagnetic selection techniques using CD14, CD31, and CD34 markers. Cells are cultured on fibronectin-coated plates in endothelial medium supplemented with various “endothelial” growth factors such as IGF-1, VEGF, FGF, and EGF (3,5,25,35). eEPCs obtained after 1 week in culture under these conditions express high levels of the monocytic marker CD14. eEPCs do not proliferate, consistent with their end-differentiated state, but secrete potent proangiogenic and antiapoptotic factors such as VEGF, IGF-1, and HGF (5,26). Furthermore, several studies have demonstrated that recruited monocytes/macrophages regulate angiogenesis in the ischemic tissue (5,28).
We hypothesized that unfractionated PBMCs cultured in suspension (S-PBMCs) are equivalent to eEPCs cultured under regular conditions and that their intravenous injection could preserve cardiac function following MI. In an attempt to simplify the culture medium, we first demonstrated that fibronectin and IGF-1 are required for differentiation of eEPCs while other growth factors (e.g., VEGF, EGF, and FGF) were not essential. Intravenous injection of S-PBMCs in NOD SCID mice reduced cardiac fibrosis, enhanced capillary density, and improved % LVFS in a manner similar to eEPCs. These data suggest that injected CD14+ cells played a major role in preserving cardiac function following MI. Normalizing the number of CD14+ for the in vivo studies increases CD34+ cell population as well. CD34+ contained in the S-PBMC mix may indeed play a significant role in preserving cardiac function. Previous publications reported that human peripheral blood CD34+ cells can transdifferentiate into cardiomyocytes, mature endothelial cells, and smooth muscle cells in vivo (38). Flow cytometry analysis of eEPCs and S-PBMCs indicate that 1.3 × 105 or 1.4 × 106 CD34+ cells were contained in the eEPCs and in the S-PBMCs preparation, respectively. However, injection of eEPCs and S-PBMCs into NOD SCID mice after MI improved % LVFS in a similar manner (44.2% and 43.3%, respectively), indicating that monocytes play a critical role in preserving cardiac function as well. The concept of monocytes being able to contribute to angiogenesis is not novel. As early as 2003, Urbich and colleagues showed that EPCs have distinct monocytic features, and EPCs can be cultured from CD14+ cells. In vivo, monocytes were shown to be able to contribute to angiogenesis as EPCs (27). Mononuclear cells other than EPCs contained in the PBMCs mix such as NK cells (CD56+ cells) and lymphocytes (CD4+ and CD8+ cells) can contribute as well to ischemic neovascularization by secreting angiogenic cytokines.
Treatment with EPCs has been reported to enhance ischemic angiogenesis in both preclinical and clinical studies (4,11,15,18,34). As the results of large-scale clinical trials become available, it is essential to identify ways to standardize and optimize the use of these cells, thereby providing clinicians with innovative personalized cell therapy tools to promote preservation of ischemic tissue. The ease by which the cells can be obtained and the cost of cell preparation will influence their translation to the clinic. It is therefore important to optimize the culture method (reducing processing steps, costs and time) to bring such therapy from bench to bedside affordably. Cell culture bags represent an optimal way to culture eEPCs as they allow reducing the time required for cell preparation and related cost. It is very hard to compare with precision the cost of such methods as they are different from one laboratory to another, and also because price of materials fluctuates over time. However, we have estimated that preparation of S-PBMC requires significantly less time and is on average eightfold less expensive than preparation of eEPCs produced under adherence conditions (Tables 1 and 2). Culturing cells in suspension reduces the stress due to trypsinization and centrifugations as well as the risks of contamination as the cells are maintained in a closed system. Bags used in this study were sealed, resistant, inert, breathable, and well suited for use in centrifuges. In addition, the method we propose employs on average 280 ml of culture medium whereas the regular eEPC culture technique employs 11,480 ml of culture medium to process a 100-ml apheresis (Tables 1 and 2). The regular eEPC culture method requires 10% (1148 ml) of human serum, making the use of autologous serum impossible. The volume of culture medium is therefore a critical constraint for the use of autologous serum obtained from donor plasma. This is now feasible using our method as 95% less culture medium is required than with the regular technique.
Estimation of the Cost and Time Required for Culture of eEPCs Under Adherence Conditions as Currently Performed in Preclinical and Clinical Trials

Data are reported as mean ± SD (n = 3). PBMCs, peripheral blood mononuclear cells; eEPCs, early endothelial progenitor cells.
Estimation of the Cost and Time Required for Culture of S-PBMCs in Suspension

Data are reported as mean ± SD (n = 3). IGF-1, insulin-like growth factor-1; S-PBMCs, PBMCs cultured in suspension in bags.
Previous studies demonstrated that monocytes/macrophages can be cultured under nonadherence conditions. VanDer Meer et al. were the first to culture monocytes in suspension. These authors showed that the morphology, differentiation potential, and chemotaxis of monocytes maintained in Teflon culture bags were similar to those of cells cultured under adherence conditions (20,36). Hydrophobic culture bags were suggested for the preparation of clinical monocyte-derived dentritic cells used as antitumor cell therapy as they are generated, antigen loaded, and matured in a closed system (10).
We have now shown in this study a novel and refined eEPC culture method using unfractionated PBMCs typically found in a standard apheresis collection that is well adapted to the safety requirements of cell therapy. The protocol here developed utilizes components that are commercially available (produced under GMP conditions) and approved by regulatory agencies in several countries. We propose that intravenous S-PBMCs administration for acute and chronic vascular insufficiency syndromes may improve clinical outcomes.
Footnotes
Acknowledgments
We would like to thank Dr. Nicoletta Eliopoulos (Laboratory Director at the Jewish General Hospital Cell Processing Center-JGHCPC, Montreal, QC, Canada) and Shala Yuan (Laboratory Manager at JGHCPC) for their assistance to estimate the cost and time required for eEPC preparation. This work was supported by Grants 72553 (to J.G.) and 82790 (to E.L.S.) from the Canadian Institutes for Health Research (CIHR), a Canada Research Chair (CRC, to E.L.S.) from the CRC/CIHR program of the Government of Canada, Canadian Stem Cell Network (CSCN). and Fonds de Recherche en Santé du Quebec (FRSQ). Manaf Bouchentouf is the recipient of a FRSQ scholarship.