
Abstract
While therapeutic cell transplantations using progenitor cells are increasingly evolving towards phase I and II clinical trials and chemically defined cell culture is established, standardization in biobanking is still in the stage of infancy. In this study, the EU FP6-funded CRYSTAL (CRYo-banking of Stem cells for human Therapeutic AppLication) consortium aimed to validate novel Standard Operating Procedures (SOPs) to perform and validate xeno-free and chemically defined cryopreservation of human progenitor cells and to reduce the amount of the potentially toxic cryoprotectant additive (CPA) dimethyl sulfoxide (DMSO). To achieve this goal, three human adult progenitor and stem cell populations—umbilical cord blood (UCB)-derived erythroid cells (UCB-ECs), UCB-derived endothelial colony forming cells (UCB-ECFCs), and adipose tissue (AT)-derived mesenchymal stromal cells (AT-MSCs)—were cryopreserved in chemically defined medium supplemented with 10% or 5% DMSO. Cell recovery, cell repopulation, and functionality were evaluated postthaw in comparison to cryopreservation in standard fetal bovine serum (FBS)-containing freezing medium. Even with a reduction of the DMSO CPA to 5%, postthaw cell count and viability assays indicated no overall significant difference versus standard cryomedium. Additionally, to compare cellular morphology/membrane integrity and ice crystal formation during cryopreservation, multiphoton laser-scanning cryomicroscopy (cryo-MPLSM) and scanning electron microscopy (SEM) were used. Neither cryo-MPLSM nor SEM indicated differences in membrane integrity for the tested cell populations under various conditions. Moreover, no influence was observed on functional properties of the cells following cryopreservation in chemically defined freezing medium, except for UCB-ECs, which showed a significantly reduced differentiation capacity after cryopreservation in chemically defined medium supplemented with 5% DMSO. In summary, these results demonstrate the feasibility and robustness of standardized xeno-free cryopreservation of different human progenitor cells and encourage their use even more in the field of tissue-engineering and regenerative medicine.
Keywords
Introduction
With the introduction of cell- and tissue-based therapies, reliable cryopreservation and biobanking have become progressively important (11,13,34,47) since a variety of preclinical and clinical studies already showed the promising therapeutic value of various stem and progenitor cell transplantation protocols (24,27,38). While cryopreservation of hematopoietic stem cells for clinical applications is routinely performed (1,50), cryopreservation protocols for other emerging progenitor cell populations are under development and still need to demonstrate their practical effectiveness (30,37).
In this study, we focus on the cryopreservation of umbilical cord blood (UCB)-derived erythroid cells (UCB-ECs), UCB-derived endothelial colony forming cells (UCB-ECFCs), and adipose tissue (AT)-derived mesenchymal stromal cells (AT-MSCs) in view of their clinical potential (20,43,45,46,55). Although significant improvements in cryopreservation of these cell populations have been achieved at the laboratory level, the translation with respect to the derivation, enumeration, and storage for clinical applications [e.g., to generate cells in compliance with current Good Manufacturing Practices (cGMP)] is still in the state of infancy (12,47). One concern is the development and usage of cGMP grade reagents free of animal serum for the entire process (7,39,56).
In cell-based therapies, the risk of transmitting potentially infectious agents to humans is a real concern. Especially fetal bovine serum (FBS) used in the cryopreservation media is difficult to remove during washing and any residue left can trigger adverse reactions in patients receiving cell infusions or transplants (3,16). FBS is a natural mix of growth factors, hormones, and nutrients supporting cell survival and proliferation, but also contains many uncharacterized components that might display the following drawbacks. i) Variability in serum composition makes standardized cell preparation and cultivation, which is a prerequisite for clinical transplants, very complex and not reproducible. For example, it was reported by the Cancer Vaccine Consortium of the Sabin Vaccine Institute (CVC/SVI) that the serum choice among their participants was responsible for suboptimal performance in one of their international Enzyme Linked Immuno Spot Technique (ELISPOT) proficiency panels (21). ii) Development of antibodies to FBS components has been demonstrated to occur in vivo (32,44,49). Metabolic uptake of nonhuman sialic acid Neu5Gc by cultured human cells from the FBS in culture medium, against which many humans possess circulating antibodies, might lead to complement activation and cell death in vivo after transplantation (33). iii) The potential risk of transmitting known or unknown animal pathogens from FBS into the human recipient might lead to for example bovine spongiform encephalopathy virus to infect humans with a variant of Creuzfeld-Jakob disease (6,52).
For all these reasons, cell preparations in the presence of animal sera are under increased scrutiny by regulatory authorities (25). Moreover, in various countries it is difficult to pass treatment protocols for cellular transplantation therapy that rely on FBS-expanded cells. Guidelines have been put in place to reduce the risk of possible transmission from cell-based products into humans and involve restriction of the sources of FBS, as well as guidelines for production and handling (3,19). Nevertheless, the best approach is to remove possible harmful sources of contamination for cell-based therapies and products, which means the removal of animal-derived serum from the entire production process.
This issue has prompted the successful development of serum-free and xeno-free isolation, proliferation, and maintenance media in recent years. However, the elimination of FBS should not be restricted to the cultivation of cells, but should also be applied to the storage of cell stocks. Many infectious agents like bacteria and viruses are capable of surviving adverse conditions like vapor-phase liquid nitrogen temperatures (–196°C) that are routinely used for the storage of cell stocks (4,18). Vitrification in open straws has been successfully and extensively used (40); however, it is difficult to maintain the liquid nitrogen and the cells in this open system sterile. GMP-compliant straws, which are essential for clinical grade vitrification, allow cryopreservation only for a small amount of cells. In addition, it will be imperative to develop a less toxic or even nontoxic CPA for cryopreservation and biobanking (35). The frequently used CPA DMSO is potentially toxic at the used concentrations, which complicates use of cryopreserved cells in patients (22,41,53). Furthermore, addition and removal of a CPA is a complex process associated with a potentially detrimental osmotic shock to the cells (31).
Several serum-free cryopreservation media and methods have been developed and distributed on the market as GMP-compliant or GMP-amenable products (15,17, 29,51). IBMT-medium, used in the present study, was specifically designed to compensate for a cell's changing environment at low temperatures under xeno-free and chemically defined conditions (14). Our aim in this study was to validate this xeno-free and chemically fully defined cryopreservation medium as a next step for the complete xeno-free sequence of in vitro cell processing. Additionally, we aimed to investigate the effect of a reduced addition of DMSO on cell physiology following cryopreservation.
Materials and Methods
Isolation and Culture of Progenitor Cell Populations
UCB-ECs were cultured as previously described (10). Briefly, cord blood was collected following elective cesarean sections (gestational age: week 38–40) and with informed consent of the mothers, according to institutional guidelines of the Medical University of Vienna, Austria. The blood was diluted 1:2 with PBS, and mononuclear cells (MNCs) were separated from erythrocytes by density separation on a Ficoll cushion. The mononuclear cells were washed twice with PBS and then resuspended in StemSpan Serum-Free Expansion Medium (SFEM; StemCell Technologies) supplemented with 2 U/ml erythropoietin (Erypo, Jannsen-Cilag, 10,000 U/ml), 100 ng/ml Stem Cell Factor (SCF), 1 × 10–6 M dexamethasone (Dex), 40 ng/ml insulin-like growth factor-1 (IGF-1), and 20 μg/ml of a cholesterol-rich lipid mix (all from Sigma) at a concentration of approximately 10 × 106 cells/ml. Partial medium changes were performed daily, and adherent cells were removed by daily transfer to fresh culture dishes. Outgrowth of erythroid progenitor cells was monitored by staining of cytospin preparations according to May-Gruenwald-Giemsa and by analyzing the CASY cell counter (Schaerfe, Reutlingen) profiles. As soon as erythroid cells start to dominate the culture (day 6–10), the cell concentration was adjusted to 2 × 106 cells/ml and maintained at this value by daily partial medium changes. At day 14, more than 95% show the characteristics of erythroid progenitor cells in hematological staining, cell diameter, and flow cytometry (>95% of the cells are positive for CD71 and CD36).
UCB-ECFCs were cultured as previously described (56). Briefly, at term UCB units were collected immediately after elective cesarean section from women without signs of infection (gestational age: week 38–40). Informed consent of the mothers was obtained, according to institutional guidelines of the University Hospital Zurich, Switzerland. The citrate-phosphate-dextrose-adenine anticoagulant-1 bag system (MacoPharma, Mouvaux, France) was used for blood collection and the units were processed within 1 h. For MNC isolation, a UCB unit was diluted 1:2 with phosphate-buffered saline (PBS) containing 2 mM ethylenediaminetetraacetate (EDTA; Sigma, Buchs, Switzerland), then processed by Ficoll-Hypaque solution (Biochrom AG, Berlin, Germany) gradient centrifugation at 500 × g for 30 min at room temperature. MNCs were removed from the interphase and washed once with cold PBS/EDTA, pelleted, then resuspended in endothelial cell growth medium (EGM-2, Lonza) and plated on fibronectin-coated (Biomedical Technologies Inc., Stoughton, MA) T75 tissue culture plates at a density of 5 × 105/cm2. After overnight incubation, nonadherent cells were removed and fresh endothelial medium was added. Medium was refreshed every 7 days. First cobblestone-like colonies appeared between days 12 and 20 after plating. The colonies were enumerated and then harvested by accutase (Sigma) treatment and replated at seeding densities of 2–4 × 103 cells/cm2 in T75 flasks, then cultured till confluence and serially passaged.
AT-MSCs were cultured as previously reported (57). Briefly, an adipose tissue aspirate (obtained from liposuction after plastic surgery at the plastic surgeon cabinet with informed consent of the donor according to Belgian law) was separated into two fractions: a floating adipose fraction [containing processed lipoaspirate cells (PLA)) and a denser fluid (liposuction aspirate fluid cells (LAF)]. Next, 10 ml of the PLA fraction was digested with a 0.15% collagenase solution and centrifuged for 10 min at 800 × g. Cells were then resuspended in CCM, consisting of low glucose Dulbecco's modified essential medium (DMEM) supplemented with 15% fetal bovine serum (FBS) + 100 U/ml penicillin + 100 μg/ml streptomycin (all from Invitrogen), and filtered through a 40-μm mesh filter, before final plating in T75 culture flasks in CCM. Culture medium was refreshed every 3–4 days until 80% confluence. For initial subcultivation, cells were detached by trypsin/EDTA treatment and replated in T75 flasks at a density of 3 × 103 cells/cm2. Following establishment, AT-MSC cultures were expanded in T75 culture flasks in CCM. For splitting, cells were detached by trypsin/EDTA treatment and replated in T75 flasks at a density of 1.6 × 103 cells/cm2 in 20 ml CCM.
Potential mycoplasma contamination was excluded by PCR using either a mycoplasma detection kit (Miner-vaBiolabsassay, Berlin, Germany) or performed as previously described (5).
Immunophenotyping
Immunophenotyping of UCB-ECs (10), UCB-ECFCs, and AT-MSCs (56) was performed as previously described. Briefly, cells were detached and antibody (Ab) staining was performed on live cells. Primary Abs were directly labeled with fluorescein thiocyanate (FITC), phycoerythrin (PE), or tetramethylrhodamine isothiocyanate (TRITC); in the case of unlabeled primary Abs, dye-labeled goat or rat anti-mouse IgG1, IgG2, or IgG3 (all from Becton Dickinson, Franklin Lakes, NJ, USA) were used as secondary Abs. For staining of intracellular von Willebrand factor (vWf), cells were permeabilized with 0.1% Triton X-100 for 3 min.
The following Abs were used for UCB-ECs: anti-CD3 (clone UCHT1), anti-CD34 (clone 581), anti-CD29 (clone K20), anti-CD81 (clone JS64) from Beckman-Coulter; anti-CD14 (clone MϕP9), anti-CD36 (clone CB38), anti-CD44 (clone G44-26), anti-CD45 (clone HI30), anti-CD71 (clone M-A712), anti-CD117 (clone YB5.B8), anti-CD235A (clone GA-R2/HIR2) all from BD Biosciences; and anti-CD133 (clone 293C3) from Miltenyi Biotec.
The following Abs were used for UCB-ECFCs: anti-CD45 (clone 5B1), anti-CD133 (clone AC133) from Miltenyi Biotec; anti-CD105 (clone MEM-226) from Abcam; anti-CD31 (Part No. 2003788), anti-CD54/ICAM-1 (Part No. 2003785), anti-von Willebrand factor (vWF, Part No. 2003787), anti-CD146 (Part No. 2003789) from Chemicon International; anti-KDR (89106) from R&D Systems; and anti-CD14 (clone M5E2), anti-CD62E/E-selectin (clone 68-5H11), anti-CD117 (clone YB5.B8), anti-CD144 (clone 55-7H1) from Becton Dickinson.
The following Abs were used for AT-MSCs: anti-CD11b (clone ICRF44), anti-CD79a (clone HM47), from BioLegend; anti-CD34 (clone AC136), anti-CD45 (clone 5B1) from Miltenyi Biotec; anti-CD14 (clone M5E2), anti-CD44 (clone G44-26), anti-CD73 (clone AD2), anti-CD90 (clone 5E10), anti-CD166 (clone GHI/61), anti-MHC-I (clone G46–2.6), anti-MHC-II (clone G46-6) from Becton Dickinson; and anti-CD105 (clone MEM-226) from Abcam.
Unspecific isotype Abs were used as controls. Cells were fixed in 4% buffered formalin and analyzed with a flow cytometer (FACSCalibur, Becton Dickinson). A minimum of 104 gated events were acquired for each sample.
Functional Characterization of Fresh or Cryopreserved UCB-ECs
Terminal erythroid differentiation was induced by changing the cytokine composition in the cultivation medium as described (26). Cells were washed once in PBS and seeded in StemSpan SFEM supplemented with the following factors: 10 U/ml erythropoietin, insulin 4 × 10–4 IE/ml (Actrapid, NovoNordisk 100 IE/ml), 10–6 M T3 thyroid hormone, 3 × 10–6 M mifepristone (RU486), 3% human serum (type AB, male) and 1 mg/ml transferrin (all from Sigma). The factors were replenished by daily partial medium changes. Differentiation was followed by monitoring the decrease in cell size with the CASY counter, by benzidine staining of cytospin preparations, and by a colorimetric hemoglobin assay as described in Dolznig et al. (10).
Functional Characterization of Fresh or Cryopreserved UCB-ECFCs
In order to visualize capillary formation, UCB-FCs were seeded at 5 × 103 to 2 × 104 cells/well of a 96-well tissue culture plate precoated with 50 μl Matrigel (Becton Dickinson). Capillary formation was monitored over the course of 8 h with an inverted phase microscope (DMIL microscope; Leica, Glattbrugg, Switzerland) equipped with a DML digital camera and LeicaQ Win Image Analysis software (Leica Imaging System Ltd, Cambridge England, UK). HUVEC cells were used as positive control in this assay. In order to monitor endothelial cell activation, UCB-ECFCs were incubated with 10 ng/ml of recombinant human tumor necrosis factor-α (TNF-α; Life Technologies) for 4 h at 37°C. Next, upregulation of CD62E and CD54 was determined by flow cytometry using PE-labeled anti-CD62E and anti-CD54 antibodies (Becton Dickinson).
Functional Characterization of Fresh or Cryopreserved AT-MSCs
AT-MSCs were tested in vitro for their differentiation potential into three mesodermal lineages. Differentiation was induced by culture in commercial differentiation media: AdipoDiff, ChondroDiff, and OsteoDiff Induction Media (Miltenyi Biotec GmbH, Germany). Control cultures were grown in NH expansion medium (Miltenyi Biotec GmbH, Germany). For adipogenic differentiation, AT-MSCs were cultured at 5 × 104 cells/well in 12-well plates (TPP, Switzerland) in AdipoDiff medium for 3 weeks, with fresh media added every 48 h. Oil Red O staining was used to visualize lipid droplets. Cells were washed twice with PBS, fixed in precooled methanol for 5 min, washed, and then incubated with 0.5% Oil Red O (Sigma-Aldrich AG, Switzerland) in isopropanol for 20 min at room temperature. Finally, stained cells were washed and analyzed microscopically. Chondrogenic differentiation was performed under micromass condiin ChondroDiff medium. Briefly, 5 × 106 AT-MSCs pelleted by 5-min centrifugation at 300 × g. The micromass was kept in ChondroDiff medium for 24 days, with fresh media added every 48 h. The pellets were fixed in 10% formalin, and then embedded in paraffin. Staining for glycosaminoglycans was performed on deparaffinized 5-μm sections alone or in combination with periodic acid-schiff (PAS), which stains glycogen. For detection of glycoconjugates, sections were immersed in a solution of 1% Alcian Blue dissolved in 3% aqueous acetic acid (pH 2.5) for 10 min. The slides were washed and immersed in Schiffs reagent for 10 min. Subsequently, the sections were washed for 10 min, counterstained with Weigerts hematoxylin, dehydrated in graded ethanol, cleared in xylene, and mounted in resinous medium. For osteoenic differentiation, 3 × 104 AT-MSCs were seeded in 12-well plates (TPP) and maintained in OsteoDiff induction medium for 3 weeks and fresh medium was added every 48 h. After 3 weeks, cells were fixed in 70% ethanol. Alkaline phosphatase production of the cells was evaluated by BCIP-NBT assay (GIBCO). Briefly, the BCIP-NBT solution was prepared and the cells were stained as described in the manufacturer's protocol. Finally, stained cells were washed and analyzed microscopically.
Cryomedia
For the experiments the following cryomedia were used: standard cryomedium (FBS-10:90% FBS + 10% DMSO), serum-/xeno-free chemically defined cryomedium 10 (CM-10: IBMTSTEM + 10% DMSO), cryomedium 5 (CM-5: IBMTSTEM + 5% DMSO), cryomedium 2 (CM-2: IBMTSTEM + 2% DMSO), or cryomedium 0 (CM-0: IBMTSTEM without DMSO; IBMT-media, Fischer Procryotect, Zurich, Switzerland, licensed by Fraunhofer IBMT, St. Ingbert, Germany).
Cryopreservation of Progenitor Cell Populations
Following harvesting of UCB-ECs, UCB-ECFCs and AT-MSCs, cells were washed twice with PBS and resuspended at a concentration of 1 × 106 cells/ml in either FBS-10, CM-10, or CM-5. Next, 1.5-ml aliquots were transferred to ice-cooled cryovials and a 0.2-ml sample was directly removed from each tube to perform cell count and viability analysis prefreezing. Then, cell samples were cooled in a standard freezing container (“Mr. Frosty,” Nalgene, USA) to −80°C at −1°C/min. After ≥ 2 h, samples were transferred to a 1 N2 tank where they were kept for at least 24 h prior to thawing. For thawing of cryopreserved samples, cryovials were placed in a 37°C water bath for rapid thawing until almost no ice was detectable. Directly after thawing, a 0.2-ml sample was removed from each tube to perform cell count and viability analysis postfreezing (= cell recovery analysis). Next, the cell suspension was diluted in 15 ml of fresh culture medium and incubated for 10 min at 37°C. Following centrifugation, cells were resuspended in 6 ml prewarmed culture medium and subcultured in duplicate culture. At different time points postplating (24 and 48 h for UCB-ECs, 48 and 96 h for UCB-ECFCs and AT-MSCs), cells (including supernatant) were harvested to perform cell count and viability analysis (= cell repopulation analysis). Each cell sample was counted in triplicate using a hemocytometer or automatic cell counter and cell viability was determined by propidium iodide exclusion during flow cytometric analysis.
Scanning Electron Microscopy (SEM) of Cryopreserved UCB-ECs, UCB-ECFCs, and AT-MSCs Postthaw
After cryopreservation in CM-0, CM-2, CM-5, CM-10, or FBS-10 and thawing, cell samples were washed in PBS, fixed in 2% glutaraldehyde in sodium cacodylate buffer, and kept at 4°C for at least 1 day. Next, samples were washed in PBS and seeded for at least 2 h in PBS or culture medium without FBS on ethanol-treated and dried aluminum foils. Samples were prepared for SEM according to standard procedures: washed in water, dehydrated in increased serial ethanol, critical point dried, and coated with heavy metals. Samples were examined with secondary electron (SE) or backscattered electron (BSE) mode in FESEM XL30 (Phillips, USA) at 10 kV accelerating voltage and a 10-mm working distance.
Multiphoton Laser-Scanning Cryomicroscopy (MLSCM) of Cryopreserved UCB-ECFCs and AT-MSCs
The multiphoton laser-scanning microscope LSM510-Meta-NLO (Zeiss, Germany) was used to improve spatial resolution, penetration depth, and photo quality of cell observation during cryopreservation (9). Cells (1.5 × 104–3.8 × 104)/cm2 were cultivated for at least 24 h on Thermanox coverslips (Nunc, Germany) with a diameter of 13 mm. The cytoplasm of cells was stained with 2 μg/ml calcein (Invitrogen, UK), the nucleus with 10 μg/ml Hoechst 33,342 (Sanofi-Aventis, Frankfurt, Germany) by incubating for 15 min at 37°C. After staining, cells were incubated for 5 min at 4°C in CM-0, CM-2, CM-5, CM-10, or FBS-10. Afterwards, cells were placed in a liquid nitrogen-cooled cryostage with computerized temperature feedback (temperature range +600°C down to −196°C, MDS 600, Linkam Scientific Instruments, UK). The cryostage is connected to a temperature controller (TMS 94), a liquid nitrogen pump (all Linkam), and a Dewar for liquid nitrogen supply. Stained samples were frozen at 1°C/min from 4°C to −80°C and thawed with 60°C/min to 20°C. For excitation a turnkey Ti: Sapphire femtosecond (fs) laser system (Mai Tai Broadband, Spectra-Physics, CA), which delivers 120 fs laser pulses at a repetition rate of 80 MHz, was used with a central wave length of 800 nm. Images were collected and analyzed using the Zeiss LSM image browser software, version 3,5,0,376.
Statistical Analysis
For in vitro cell recovery, repopulation, and differentiation studies, results are expressed as mean ± SD and comparisons were validated using Student's t-test with a value of p < 0.05 considered to be statistically significant using GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, CA, USA). For recovery and repopulation data, results are presented by day-per-day comparisons between the different groups with a value of p < 0.006 (for 0.05 critical significance level according to Bonferroni correction) considered to be statistically significant. The individual effects of FBS-10, CM-10, and CM-5 with respect of UCB-EC volume reduction and HB content during differentiation postthaw in function of time were validated with values of p < 0.002 and p < 0.003, respectively (for 0.05 critical significance level according to Bonferroni correction) considered to be statistically significant.
Results
Derivation and Characterization of Primary Cell Cultures
In order to perform comparative cryopreservation experiments, three distinct ex vivo cultured human adult primary progenitor cell (PC) populations were initiated and consequently tested for mycoplasma contamination, characterized by morphology and immunophenotype: i) UCB-ECs (n = 3 individual donors), ii) UCB-ECFCs (n = 3 individual donors), and iii) adipose tissue AT-MSC (n = 6 individual donors). All three PC populations were mycoplasma free, displayed uniform morphology (Fig. 1A, C, E) and characteristic immunophenotype (Fig. 1B, D, F), according to internationally well-established and accepted criteria (42,48,54). In addition, functional studies were performed to further characterize the established PC populations (Fig. 2). For UCB-ECs, in vitro differentiation studies into red blood cells indicated a uniform decrease in cell volume and increase in hemoglobin content as demonstrated by cytospin analysis (Fig. 2A), CASY-mediated cell volume measurement (Fig. 2B), and hemoglobin determination (Fig. 2C). Proliferating UCB-ECs (Fig. 2A, upper panel) consist of undifferentiated blasts with a cell size of 800 fl. Twenty-four hours after induction of differentiation, the cell size started to decrease to a final volume of 200 fl (Fig. 2B) with concurrent accumulation of hemoglobin detected by benzidin staining (Fig. 2A, middle and lower panel) and colorimetric assay (Fig. 2C). For UCB-ECFCs, capillary formation on Matrigel (Fig. 2D) and upregulation of ICAM-1 (Fig. 2E) and E-selectin (Fig. 2F) upon TNF-α stimulation demonstrated functional endothelial cell properties. For AT-MSCs, in vitro differentiation studies indicated their broad spectrum of differentiation potential towards cell types of mesodermal origin. Differentiation into adipocytes (Fig. 2G), osteocytes (Fig. 2H), and chondrocytes (Fig. 2I) could be demonstrated upon specific stimulation.

Isolation and characterization of UCB-ECs, UCB-ECFCs, and AT-MSCs. Morphology (A, C, E) and immunophenotype (B, D, F) of umbilical cord blood (UCB)-derived erythroid cells (UCB-ECs, A, scale bar: 20 μm, and B), UCB-derived cobblestone-like endothelial colony forming cells (UCB-ECFCs, C, scale bar: 100 μm, and D), and adipose tissue (AT)-derived mesenchymal stromal cells (AT-MSCs, E, scale bar: 100 μm, and F). Immunophenotyping was performed as described in Materials and Methods.

Functional properties of UCB-ECs, UCB-ECFCs, and AT-MSCs. Functional characterization of cultured UCB-ECs (A–C), UCB-ECFCs (D–F), and AT-MSCs (G–I). For UCB-ECs, in vitro differentiation studies into red blood cells indicated a uniform decrease in cell volume and increase in hemoglobin content as demonstrated by cytospin analysis (A, scale bar: 20 μm), CASY-mediated cell volume measurement (B, black circles indicate cells in the presence of maintenance and white circles in differentiation medium), and colorimetric hemoglobin determination (C, black circles indicate cells in the presence of maintenance and white circles in differentiation medium). For UCB-ECFCs, formation of capillary-like structures on Matrigel (D, scale bar: 100 μm) and upregulation of ICAM-1/CD54 (E) and E-selectin/CD62E (F) following TNF-α stimulation (isotyp-contol Abs in gray, unstimulated in bold line, and stimulated in dashed line) demonstrated functional endothelial cell properties. For AT-MSCs, in vitro differentiation studies indicated their tripotential differentiation capacity to produce adipocytes (G, Oil Red O staining, scale bar: 40 μm), osteocytes (H, BCIP-NBT assay, scale bar: 40 μm), and chondrocytes (I, PAS-staining, scale bar: 100 μm). Insets indicate unstimulated control samples.
Selection of a Serum- and Xeno-Free Chemically Defined Cryopreservation Medium for UCB-ECs, UCB-ECFCs, and AT-MSCs
First, a prospective analysis of commercial serum-free cryomedia was performed (Table 1, commercial cryomedia) and four media were selected for potential studies: IBMTPLUS, IBMTSTEM, Cryostor CS-10, and Cryopan II. These media all contained 10% DMSO with the exception of IBMTSTEM, which only contains 5% DMSO. Osmolarities of the media containing 10% DMSO was in the range of 2,100–2,500 mOsmol, and freezing points varied between −26.8°C and −33.4°C. Only three media, IBMTPLUS, IBMTSTEM, and Cryostor CS-10, fulfilled the criterion of being free of xenogenic compounds. The IBMT media utilize the compound Pluronic F-68 at a concentration of 1% in a chemically fully defined, although not disclosed, mixture (14). As initial tests with this cryomedium were very promising, we decided to use this solution for all further comparisons to serum-based media. Additionally, we used DMSO-free formulation of IBMTSTEM and customized the DMSO concentration for our tests. The physical properties of three different customized media (IBMTSTEM with 0%, 5%, and 10% DMSO) were analyzed (Table 1, CM-0, CM-5, and CM-10). This analysis clearly showed that the osmolarity strongly increases and the freezing point decreases with increasing DMSO concentrations. These customized media were used for all subsequent experiments. Furthermore, all comparisons were made versus cryopreservation in commonly used serum-containing cryomedium (Table 1, common cryomedia: 90% FBS + DMSO).
Comparison of Physical Properties and Ingredients of Different Cryomedia

Cryomedia investigated in this study are shown in bold. ND, not determined, NA, data not available.
Serum- and Xeno-Free, Chemically Defined Cryopreservation of UCB-ECs, UCB-ECFCs, and AT-MSCs
In order to perform a comparative survival analysis between PCs cryopreserved in a chemically defined versus FBS-containing cryomedium, we used three different parameters: “cell recovery,” “cell repopulation,” and “Δ recovery and repopulation” (Fig. 3). Cell recovery is defined as the percentage of viable cells recovered following cryopreservation in chemically defined freezing or standard FBS-containing freezing medium directly postthaw compared to the number of viable cells before freezing. Cell repopulation is defined as the percentage of viable cells recovered following cryopreservation in chemically defined or standard FBS-containing freezing medium at different time points postplating of cryopreserved cells (24 and 48 h for UCB-ECs, 48 and 96 h for UCB-ECFCs and AT-MSCs). Δ recovery and repopulation is defined as the mean cell repopulation of PC cryopreserved in chemical defined medium versus PCs cryopreserved in FBS-containing medium. During each cryopreservation experiment, three cryovials were prepared for every condition (i.e., cryopreservation in standard FBS-containing medium with 10% DMSO) (Fig. 3, FBS-10), chemically defined cryopreservation medium with 10% DMSO (Fig. 3, CM-10), and chemically defined cryopreservation medium with 5% DMSO (Fig. 3, CM-5). The absolute number of viable cells in each tube were directly counted before (Fig. 3A, C, and E; dashed line indicating 100%) and after (Fig. 3A, C, and E; white bars postthaw) cryopreservation using a hemocytometer/automatic cell counter combined with FACS analysis, as described in Materials and Methods. In addition, following cryopreservation, triplicate cultures were plated and the absolute number of viable cells in each culture was determined again after 24 h (Fig. 3A; gray bars) and 48 h (Fig. 3A; black bars) of culture for UCB-ECs and after 48 h (Fig. 3C, E; gray bars) and 96 h (Fig. 3C, E; black bars) of culture for UCB-ECFCs and AT-MSCs. Figure 3 provides a representative example for a comparative analysis of cell recovery and cell repopulation for UCB-ECs (Fig. 3A), UCB-ECFCs (Fig. 3C), and AT-MSCs (Fig. 3E). The graphs on the right (Fig. 3B, D, F) provide the Δ recovery and repopulation summarized for all experiments performed with UCB-ECs (n = 8), UCB-ECFCs (n = 9, passages 5–7), and AT-MSCs (n = 15, passages 2–8) and indicate no statistical difference for CM-10 versus CM-5.
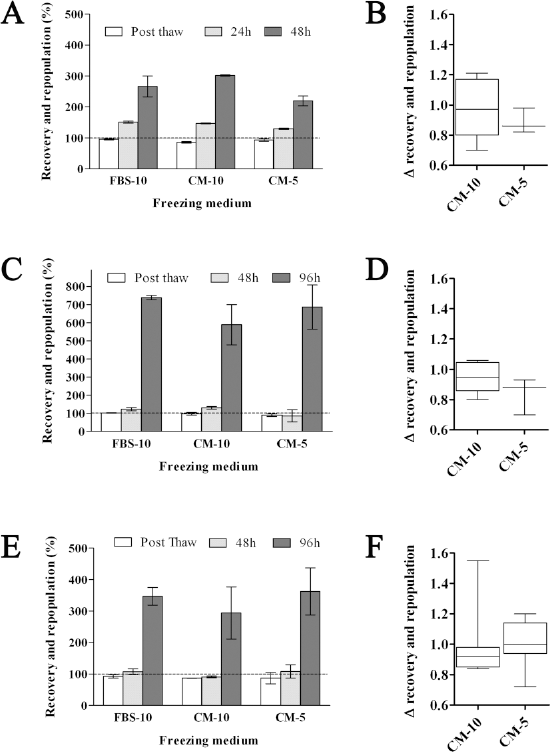
Cell recovery and repopulation of UCB-ECs, UCB-ECFCs, and AT-MSCs after serum- and xeno-free chemically defined cryopreservation. “Cell recovery,” “cell repopulation,” and “Δ recovery and repopulation” for UCB-ECs (A, B), UCB-ECFCs (C, D), and AT-MSCs (E, F) cryopreserved in FBS-10, CM-10, and CM-5. Representative example given of cell recovery provided as the percentage of viable cells recovered following cryopreservation directly postthawing compared to the number of viable cells before freezing (A, C, E; dashed line and white bars postthaw). Cell repopulation is provided as the percentage of viable cells recovered following cryopreservation at different time points postplating of cryopreserved cells [24 and 48 h for UCB-ECs (n = 3 individual donors), 48 and 96 h for UCB-ECFCs (n = 3 individual donors), and AT-MSCs (n = 6 individual donors; gray and black bars)]. Δ recovery and repopulation provided as the mean cell repopulation of PCs cryopreserved in CM-5 and CM-10 versus PCs cryopreserved in FBS-10 medium for all time points tested (B, D, F). Values are mean ± SD.
Multiphoton Laser-Scanning Cryomicroscopy of UCB-ECFCs and AT-MSCs Before, During, and After Cryopreservation
After identifying comparable cryopreservation efficiency of the chemically defined media even with reduced DMSO concentrations, we investigated the effects on membrane integrity during cryopreservation and thawing in real time. Moreover, the possibility of further DMSO reduction was analyzed. Therefore, AT-MSCs (Fig. 4A–O) and UCB-ECFCs (Fig. 4P–AD) cultured on coverslips were fluorescently labeled with calcein (green fluorescence) to stain the cytoplasm of viable cells and with Hoechst 33,342 (blue fluorescence) to counterstain the nuclei. Labeled UCB-ECFCs and AT-MSCs were covered with CM-0, CM-2, CM-5, CM-10, or FBS-10 cryomedium and placed on a temperature-controlled liquid N2-cooled cryostage, as described in Materials and Methods. The processes of ice crystallization and membrane disruption before (4°C), during (–80°C), and after (4°C) cryopreservation were examined by multiphoton laser-scanning cryomicroscopy. Cryopreservation of AT-MSCs (Fig. 4A, B, C) and UCB-ECFCs (Fig. 4P, Q, R) in CM-0 resulted in significant intercellular (arrows) and intranuclear (asterisks) ice formation at −80°C. Cryopreservation in CM-2 resulted mainly in the formation of large intercellular ice crystals for both AT-MSCs (Fig. 4E, dashed arrows) and UCB-ECFCs (Fig. 4T, dashed arrows). Following thawing after cryopreservation in CM-0 and CM-2, cytoplasm was released due to membrane disruption as indicated by fading of cytoplasm staining and an increased green background fluorescence of AT-MSC (Fig. 4C, F) and UCB-ECFC samples (Fig. 4R, U). During cryopreservation in CM-5, only limited intercellular ice crystals were observed for AT-MSCs (Fig. 4H, dashed arrows), but not for UCB-ECFCs (Fig. 4W). No intercellular or intracellular ice crystal formation was observed when cells were cryopreserved in CM-10 or FBS-10 for both AT-MSCs (Fig. 4K, N) and UCB-ECFCs (Fig. 4Z, AC). Likewise, after thawing only limited cytoplasmic loss was observed for AT-MSCs cryopreserved in CM-5 (Fig. 4I), but not for AT-MSCs cryopreserved in CM-10 and FBS-10 (Fig. 4L, O) and for UCB-ECFCs cryopreserved in CM-5, CM-10, and FBS-10 (Fig. 4X, AA, AD). Based on these data, we conclude that lack of cell recovery following cryopreservation of UCB-ECFCs and AT-MSCs in CM-0 and CM-2 (data not shown) is due to intercellular and intracellular ice crystal formation, which is not observed following cryopreservation in CM-5, CM-10, and FBS-10.

Multiphoton laser-scanning cryomicroscopy (cryo-MPLSM) of UCB-ECFCs and AT-MSCs. Multiphoton laser-scanning cryomicroscopy images of AT-MSCs (A–O) and UCB-ECFCs [P-AD before (left column, 4°C), during (middle column, −80°C), and postthaw (right column, 4°C)] in CM-0, CM-2, CM-5, CM-10, and FBS-10. Representative pictures are provided for each condition. Green fluorescence indicates calcein staining of intracellular proteins. Blue fluorescence indicates Hoechst 33,342 staining of nuclei. Scale bars: 50 μm.
Scanning Electron Microscopy (SEM) of UCB-ECs, UCB-ECFCs, and AT-MSCs Postthaw
For proving these results, SEM was used to analyze membrane integrity and structural alterations of UCB-ECs (Fig. 5A–F), UCB-ECFCs (Fig. 5G–L), and AT-MSCs (Fig. 5M–R) after cells had been cryopreserved in chemically defined cryomedia containing various concentrations of DMSO. UCB-ECs are shown before (Fig. 5A) and after (Fig. 5B–F) cryopreservation. Most of the cells had a round shape (Fig. 5A, left, arrow), although some also were elongated (Fig, 5A, left, arrowhead). Further, UCB-ECs possessed heterogeneous surface features: few cells were smooth (Fig. 5A, left, arrow), whereas most of cells had a microvillous relief (Fig. 5A, left, arrowhead) or were covered with lots of vesicles (Fig. 5A, right, arrowhead) and with short microvilli (Fig. 5A, right, arrow). After cryopreservation with CM-0 or CM-2, swelling of cells was observed, as the cell diameter increased from 7–8 μm (Fig. 5A, left) to approximately 12 μm (Fig. 5B, C, left). Signs of cell damage (e.g., disturbed cell surfaces without microvilli and numerous holes) (Fig, 5B, right, arrows) were visible on the cell surface. Cryopreservation with CM-2 resulted in improved membrane integrity (Fig. 5C). But nevertheless, any feature of cell damage ranging from compact and smooth (Fig. 5C, inset) to disturbed surface with numerous ruffles and vesicles was seen (Fig. 5C, right, arrows). Cryopreservation of UCB-ECs with CM-5 and CM-10 showed the best results (Fig. 5D, E). Thus, the form, size, and cell surface hardly displayed any differences from control preparations without cryopreservation. Some cells had a wrinkled surface with single microvilli (Fig. 5E, right, arrow), others a microvillous relief (Fig. 5D, left, arrow). On the contrary, cells frozen with FBS-10 presented various responses to cryopreservation. Some cells were covered with numerous vesicles and only few microvilli (Fig. 5F, right, arrow), whereas others had a wrinkled surface with deep disruptions (Fig. 5F, left, arrow) of the plasma membrane.

Scanning electron microscopy (SEM) of UCB-ECs, UCB-ECFCs, and AT-MSCs following serum- and xeno-free chemically defined cryopreservation. SEM images of UCB-ECs (A–F), UCB-ECFCs (G–L), and AT-MSCs (M–R) before (A, G, M) and after cryopreservation in CM-0 (B, H, N), CM-2 (C, I, O), CM-5 (D, J, P), CM-10 (E, K, Q), and FBS-10 (F, L, R). For each condition a series of two images is provided at different magnification as indicated by scale bars. Description is given in the Results section.
UCB-ECFCs are shown before cryopreservation (Fig. 5G) and postthaw (Fig. 5H–L). Cells were mostly round or oval in shape within a size of up to 30 μm covered with numerous vesicles (Fig. 5G, arrowhead) and some short microvilli (Fig. 5G, arrow). Some cells in the same sample were round with only 10 μm in diameter (Fig. 5G, right). In most SEM micrographs of UCB-ECFCs small structures (below 1 μm) could be observed both on the cell surface and the substrate surface (data not shown). Our analysis showed that as origin of these structures a contamination can be excluded. Shrinkage of cells with deep holes (Fig. 5H, arrowheads) was seen immediately after cryopreservation using CM-0. Addition of 2% DMSO resulted in smooth cell surfaces with less signs of cell damages (Fig. 5I). Smoothened ruffled membrane of UCB-ECFCs, cryopreserved in CM-5 (Fig. 5J), changed to wrinkled surface with single microvilli by cryopreservation in CM-10 (Fig. 5K) or serum-containing medium (Fig. 5L).
AT-MSCs before and after cryopreservation are presented in Figure 5M and Figure 5N–R, respectively. Cells had a size of 20–30 μm, a round or oval shape, and a surface relief with microvilli and ruffles. Some-times also vesicles were seen on the surface (Fig. 5M, arrow). As expected, cryopreservation in CM-0 significantly changed cell morphology, leading to cells covered with numerous vesicles (Fig. 5N, arrows) and small holes were seen on the cell surface (Fig. 5N, arrowhead). Addition of 2% DMSO already resulted in a visible improvement (Fig. 5O). Cells were covered with numerous microvilli (Fig. 5O, left) without any signs of cell damage, and some cells presented dense vesicles (Fig. 5O, arrows). Only minor differences were seen between CM-5 (Fig. 5P), CM-10 (Fig. 5Q), and FBS-10 (Fig. 5R). In each preparation, cells were densely covered with larger microvilli; the appearances of fine long filaments can be also noted (Fig. 5P, Q, R, arrows). Nevertheless, after cryopreservation with CM-10 some shrunken cells with cavities on the cell surface could be observed (Fig. 5Q, inset).
Functional Characterization of UCB-ECs, UCB-ECFCs, and AT-MSCs Following Chemically Defined Cryopreservation
Following establishment of chemically defined cryopreservation conditions, additional functional studies were performed to demonstrate differentiation potential of UCB-ECs (Fig. 6A–C), UCB-ECFCs (Fig. 6D–F), and AT-MSCs (Fig. 6G–I) after cryopreservation in FBS-10, CM-10, or CM-5. For UCB-ECs, no difference was observed following in vitro differentiation into red blood cells, as indicated qualitatively by a uniform change of morphology (Fig. 6A) and quantitatively by a decrease in cell volume (Fig. 6B). However, after cryopreservation in FBS-10, CM-10, or CM-5, cells, which had been cryopreserved in CM-5, showed a significant lower hemoglobin content at day 7 versus FBS-10 (5.49 ± 0.12 vs. 8.03 ± 0.15, p = 0.0002) and no difference versus CM-10 (5.49 ± 0.12 vs. 8.35 ± 0.53, p = 0.006) (Fig. 6C). For UCB-ECFCs, neither quantitative differences in the upregulation of ICAM-1 (Fig. 6D) and E-selectin (Fig. 6E) following TNF-α stimulation, nor qualitative difference in capillary formation on Matrigel (Figure 6F), were observed after cryopreservation in FBS-10, CM-10, or CM-5. For AT-MSCs, in vitro differentiation studies indicated their broad spectrum of differentiation potential from cell types of mesodermal origin following cryopreservation in CM-10. These cells displayed adipogenic (Fig. 6G), osteogenic (Fig. 6H), and chondrogenic (Fig. 6I) differentiation following specific stimulation.

Functional characterization of UCB-ECs, UCB-ECFCs, and AT-MSCs following serum- and xeno-free chemically defined cryopreservation. Functional characterization of cultured UCB-ECs (A–C), UCB-ECFCs (D–F), and AT-MSCs (G–I) post-thaw. For UCB-ECs, in vitro differentiation studies into red blood cells indicated a uniform decrease in cell volume and increase in hemoglobin content as demonstrated by cytospin analysis (A, scale bar: 20 μm), CASY-mediated cell volume measurement (B, circle FBS-10, triangle pointing up CM-10, and triangle pointing down CM-5), and colorimetric hemoglobin determination (C, circle FBS-10, triangle up CM-10, and triangle down CM-5). Values are mean ± SD, n = 3 individual donors. *p < 0.003. For UCB-ECFCs, upregulation of ICAM-1/CD54 (D) and E-selectin/CD62E (E) following TNF-α stimulation and formation of capillary-like structures on Matrigel (F, scale bar: 100 μm) demonstrated functional endothelial cell properties. Values are mean ± SD, n = 3 individual donors. For AT-MSCs, in vitro differentiation studies indicated their tripotential differentiation capacity after freeze-thaw in chemically defined cryopreservation medium to produce adipocytes (G, Oil Red O staining, scale bar: 40 μm), osteocytes (H, BCIP-NBT assay, scale bar: 40 μm), and chondrocytes (I, PAS and Alcian blue staining, scale bar: 100 μm). Insets show unstimulated control samples.
Discussion
In the present study we demonstrate the successful transition from standard serum-based cryopreservation towards a protocol using a xeno-free, chemically defined cryomedium. The straightforward use of this chemically defined cryoprotocol enables freezing of large quantities of stem and progenitor cells for both research and clinical applications. Advantageously, it does not require complex cryoequipment (e.g., programmed gradient-freezer, etc.), and it is easy to handle. Even though the tested chemically defined cryomedium has not yet undergone the official cGMP validations, all the components are cGMP compatible making clinical grade achievable.
To demonstrate that chemically defined cryopreservation is feasible not only for unique cell types or cell lines, we included three different primary cultures. i) UCB-ECs were selected as a model system for hematopoietic stem/progenitor cells. These cells offer the advantage that a large number of homogeneous cells can be generated from a single UCB unit. This cell system enabled us to investigate the effects of chemically defined cryopreservation with standardized procedures, which would not have been possible with therapeutically relevant CD34+ cells due to the low cell numbers in single UCB units. In addition, the cultivation of cells is performed in chemically defined medium and only differs from existing approaches aiming at the expansion of CD34+ cells in the choice of added cytokines and growth factors. In addition, we have also analyzed the effect of chemically defined cryopreservation on the number of viable CD34+ cells in MNCs isolated from UCB and could demonstrate similar recovery rates (manuscript in preparation). These results suggest that the proposed xeno-free cryopreservation procedure is a valid alternative for hematopoietic stem/progenitor cells. ii) ECFCs isolated from UCB were selected as a therapeutically relevant cell type, which can be isolated and expanded with high reproducibility (56). UCB-ECFCs can be used for tissue engineering of vascular grafts to treat early onset, childhood cardiovascular disease (43) and are currently being investigated as a treatment for acute myocardial infarction (45). iii) Adipose tissue-derived MSCs are considered to have a high potential for regenerative medicine. Due to the suggested ability of MSCs to differentiate into various cell types, including hepatic-like, neuron-like, chondrocyte-like, or osteoblast-like cells, they might become useful to aid the treatment of various diseases (20,46,55).
Following cell isolation and expansion, immunophenotyping and functional analysis were performed to characterize and confirm progenitor cell origin (Figs. 1 and 2). The physical properties of different commercially available and widely used cryomedia were compared and we obtained one chemically defined cryomedium without DMSO that was used in our approach (Fig. 1). Varying the DMSO concentrations in this chemically defined cryopreservation medium, we developed a SOP, which allowed us to compare cell recovery and cell repopulation between the different cultures postthaw. Since suspension cultures of UCB-ECs already exhibit a very high proliferation rate directly after thawing, these cells were assayed at the time points 24 and 48 h postthaw. After 24 h, we detected a 1.5-fold expansion, which further increased to threefold at 48 h for FBS-10, CM-10, and CM-5 (Fig. 3A). Although in some samples significant differences were detected at the first and/or first two time points (Fig. 3A), no statistically significant differences were observed between FBS-10 and the two chemically defined media combining all three tested UCB-EC isolations at all time points. When comparing CM-5 and CM-10, we observed a slightly better cell recovery with the higher DMSO concentration (Figu. 3B). Similarly, CM-10 and FBS-10 also led to a higher accumulation of hemoglobin compared to CM-5, as detected in a quantitative hemoglobin production assay during terminal differentiation of UCB-ECs (Fig. 6C). Therefore, chemically defined cryopreservation of hematopoietic cells in xeno-free cryomedium containing 10% DMSO results in a better preservation of the functional properties of the cells and might thus be preferable, although no significant differences in cell recovery and repopulation or qualitatively no membrane cryodamage could be detected.
Both UCB-ECFCs and AT-MSCs were assayed 48 and 96 h postthaw, because these adherent cell populations need more recovery time postthaw following replating and for initiation of cell division. For these cell types, both CM-5 and CM-10 performed equally well compared to FBS-10 with respect to cell recovery and cell repopulation (Fig. 3D, F). Moreover, functional assays did not reveal any quantitative (Fig. 6D, E) nor qualitative differences for UCB-ECFCs (Fig. 6F). Upon cryopreservation of AT-MSCs in CM-10 or CM-5, no qualitative differences were observed after cryopreservation in CM-10 (Fig. 6G–I). Therefore, efficient cryopreservation of UCB-ECFCs and AT-MSCs can also be achieved with a chemically defined freezing protocol.
To analyze the effects of cryopreservation at the single-cell level, we monitored the freezing process of the two adherent cell cultures, AT-MSCs and UCB-ECFCs, by MPLSM cryomicroscopy. As UCB-ECs are cultivated in suspension, they were not included for this analysis. The combination of a multiphoton laser scanning microscope equipped with a cryostage enabled us to analyze cells during freezing and thawing. Intra- and intercellular ice crystal formation and loss of membrane integrity was observed for cryomedia containing 2% or less DMSO (Fig. 4A–F, P–U). For concentrations of 5% and more DMSO, only modest or no loss of membrane integrity was observed for UCB-ECFCs (Fig. 4G–O, V–AD). For AT-MSCs, we still observed formation of intercellular ice crystals during freezing with CM-5, and also found modest loss of membrane integrity postthaw.
SEM image analysis indicated qualitatively that cryopreservation in the xeno-free cryomedium containing 5% or 10% DMSO is comparable to FBS-supplemented cryomedium with 10% DMSO. In these preparations the analyzed cells had intact surfaces with features that did not differ from the control. The appearance of shrunken cells after cryopreservation of AT-MSCs with chemically defined cryopreservation medium + 10% DMSO (Fig. 5Q, inset) can be interpreted as osmotic stress response to freezing (2,36). On the contrary, appearances of holes and disruptions on the cell surface postthaw without DMSO as well as rarely with FBS-supplemented cryopreservation medium containing 10% DMSO are signs of loss of membrane integrity (Fig. 5F, H, N) and can be interpreted as cell damages that possibly lead to cell death (8). A smoothing of cell surface by cryopreservation with chemically defined cryopreservation medium + 2% DMSO (Fig. 5C, inset, I) can be explained by two different reasons. First, it can be a feature of common stress reaction in response to freezing and thawing (23). Second, we also observed smooth cells in the control of UCB-ECs (Fig. 5A, arrow). Such smoothing of the cell surface could also be a sign of spontaneous erythroid differentiation of UCB-ECs. Therefore, further investigation is necessary by analyzing the recovery of these cells with correlative light and scanning electron microscopy, using surface markers for identification of each cell population. Interestingly, in some preparations of AT-MSCs fine fibers could be detected postthaw, corresponding, probably, to extracellular matrix (Fig. 5Q, R, arrows). This could be a sign of spontaneous differentiation of MSCs by freezing stress, accompanied with ECM molecule expression (28).
Summarizing, we have now demonstrated, using three different therapeutically relevant progenitor cell populations, the efficacy of a fully defined cryopreservation protocol with high cell recovery, cell repopulation, cell membrane integrity, and postthaw cellular functionality for three distinct PC populations. The proposed protocol is xeno free, easy to use, does not require laborious equipment, and is cGMP amenable. We have further shown that a reduced supplementation of the cell permeating CPA DMSO from 10% to 5% is feasible and generally supports cell recovery, cell repopulation, membrane integrity, and maintains cell functionality postthaw comparable to 10% DMSO. Based on our presented data, we conclude that chemically defined and DMSO-reduced cryopreservation provides a relevant solution for the cryopreservation of progenitor and stem cells for laboratory and clinical use.
Footnotes
Acknowledgments
The authors would like to thank E. Kleiner-Blatter, A. Kurmanaviciene (Department of Obstetrics, University Hospital Zurich, Zurich, Switzerland), J. Daans (Laboratory of Experimental Hematology, Vaccine and Infectious Disease Institute, University of Antwerp, Antwerp, Belgium), U. Beer, M. Parth (Department of Obstetrics and Gynecology, Medical University of Vienna, Austria) and S. Zöllner (Fraunhofer IBMT, St. Ingbert, Germany) for their excellent technical assistance, A. Huber and W. Dietrich (Department of Obstetrics and Gynecology, Medical University of Vienna, Austria) for the collection of cord blood, and René Fischer for proof reading of the manuscript. This work was financed with a grant from the European Commission (FP6-project: LSHB-CT-2006-037261 “CRYSTAL,” Cryopreservation of stem cells for human therapeutic application) to P.P., G.W., J.H., A.Z., A.K., and H.Z. J.C.S. and H.Z. declare the following conflict of interest: The IBMT medium is a medium/technology, formerly known as Filoceth, and originally developed by René Fischer (Procryotect). The technology was bought by Fraunhofer IBMT after finalization of this manuscript. IBMT medium will be in future commercialized under the license of Fraunhofer IBMT.