
Abstract
Natural CD4+Foxp3+ T regulatory (Treg) cells can promote transplantation acceptance across major histocompatibility complex (MHC) barriers, while myeloid-derived suppressor cells (MDSCs) inhibit effector T-cell responses in tumor-bearing mice. One outstanding issue is whether combining the potent suppressive function of MDSCs with that of Treg cells might synergistically favor graft tolerance. In the present study, we evaluated the therapeutic potential of MDSCs and natural Treg cells in promoting allograft tolerance in mice by utilizing immunomodulatory agents to expand these cells in vivo. Upon administration of recombinant human granulocyte-colony stimulating factor (G-CSF; Neupogen), or interleukin-2 complex (IL-2C), Gr-1+CD11b+ MDSCs or CD4+Foxp3+ Treg cells were respectively induced at a high frequency in the peripheral lymphoid compartments of treated mice. Interestingly, induced MDSCs exhibited a more potent suppressive function in vitro when compared to MDSCs from naive mice. Furthermore, in vivo coadministration of Neupogen and IL-2C induced MDSCs at percentages that were higher than those seen when either agent was administered alone, suggesting an additive effect of the two drugs. Although treatment with either IL-2C or Neupogen led to a significant delay of MHC class II disparate allogeneic donor skin rejection, the combinatorial treatment was superior to either alone. Importantly, histological assessment of surviving grafts revealed intact morphology and minimal infiltrates at 60 days posttransplant. Collectively, our findings demonstrate that concurrent induction of MDSCs and Tregs is efficacious in downmodulating alloreactive T-cell responses in a synergistic manner and highlight the therapeutic potential of these naturally occurring suppressive leukocytes to promote transplantation tolerance.
Introduction
Tumorigenesis is a complex phenomenon that has been studied utilizing animal models with the attempt to understand its pathogenesis. Once established, tumors utilize several strategies to evade recognition by the immune system (8,42). One such strategy is the recruitment of myeloid-derived suppressor cells (MDSCs) through granulocyte-macrophage colony stimulating factor (GM-CSF) and granulocyte-colony stimulating factor (G-CSF) that is produced by tumor cells (15,42). The prevalence of these suppressor cells in and around the tumor microenvironment consequently contributes to tumor escape, as several animal models have demonstrated a correlation between increased frequencies of these cells within the tumor microenvironment and either persistence or progression of tumor growth (29,33). In human studies, equivalent populations of cells have also been reported in cancers such as head and neck squamous cell carcinoma (3).
Depending on the model of investigation, MDSCs have been described as expressing various cellular markers (15). In mice, they are typically contained in the CD11b+ leukocyte subset and express varying levels of Gr-1 (Ly6G/C), a cell surface molecule with heterogeneous distribution that is used to delineate their differentiation stages (7,50). They also express F4/80 (23), and interleukin-4 receptor α (IL-4Rα) (12,16). Unlike conventional CD11b+ macrophages, these cells express low levels of major histocompatibility complex (MHC) class II, CD40, and B7.1/2 costimulatory molecules, a phenotype that is distinct from that of immature and mature antigen-presenting cells (4,41,51). The potent suppressive activity of MDSCs is revealed by in vitro studies in which they effectively inhibit the proliferation of either antigen- or mitogen-activated T lymphocytes (16,43).
As the understanding of the mechanisms by which MDSCs suppress effector immune responses is gradually unfolding, certain key proteins have been identified as crucial to their suppressive function. For example, expression of IL-4Rα is paramount to arm MDSCs with the ability to function as suppressors, with the most potent cells residing within the Ly-6C-high subset (11,16). In addition, these cells express high levels of nicotinamide adenine dinucleotide phosphate (NADPH), phagocytic oxidase (heme oxygenase-1), nitric oxide synthase (NOS2), and arginase-1, enzymes that are involved in processes that lead to the production of nitric oxide (NO), peroxynitrite, reactive oxygen species (ROS), or to the depletion of the semiessential amino acid L-arginine (9,24,25,46). The resulting metabolites affect T-cell intracellular signaling by lowering the CD3-ζ chain, blocking IL-2R activated pathways, changing the affinity of the T-cell receptor (TCR)/MHC peptide complex, or inducing apoptosis (6,13,34,36,38,40).
Although there are extensive studies on the role of MDSCs in downmodulating immunity in tumor models, little is known about their role in nontumor settings. While it is well established that natural CD4+Foxp3+ Treg cells are efficacious at promoting tolerance across MHC barriers in bone marrow and solid tissue transplantation settings, there are scant data about the role of MDSCs in modulating time and/or severity of graft rejection. In recent reports, MDSCs were implicated in animal models of kidney allograft tolerance (12) and were also found to delay rejection of antigen-mismatched skin grafts (10,53). Given their potent inhibitory effect in tumor-bearing mice, one outstanding issue is whether combining the potent suppressive function of both natural CD4+Foxp3+ Treg cells and MDSCs might favor transplantation tolerance. Thus, the present study was designed to evaluate the therapeutic potential of MDSCs and natural Treg cells in promoting allograft tolerance by utilizing immunomodulatory agents to expand these cells in vivo.
Materials and Methods
Mice
C57BL/6, BALB/c, C3H/HeJ, and B6 (C)-H-2-AB1bm12/KhEgj were obtained from Jackson Laboratory (Bar Harbor, ME). All experiments were approved by the Institutional Animal and Use Committee (IACUC) at the University of Miami and performed following the IACUC guidelines.
Antibodies and Fluorescent Activated Cell Sorter (FACS) Analysis
FITC-conjugated monoclonal antibodies (mAbs) to CD4 (Gk1.5), Gr-1 (RB6–8C5), CD40 (HM40–3), CD80 (GL1), IA/E (NIMR-4), F4/80 (BM8), phycoerythrin (PE)-conjugated mAbs to Gr-1 (Ly6C/6G), CD11b (M1/70), CD124, peridinin chlorophyll protein complex (PerCP)-anti Gr-1 (RB6–8C5), allophycocyanin (APC)-conjugated CD11b (M1/70), and CD25 (PC61) were all purchased from BD Pharmingen (San Diego, CA). PE-anti-Foxp3 (FJK16s) was obtained from eBiosciences (San Diego, CA) and was used in intracellular FACS analysis following the manufacturer's instructions. FACS analysis was performed using FACs Calibur and CellQuest software. For cultured cells, 7AAD dye was utilized to exclude dead cells from the analysis. Typically 30,000–50,000 events were collected per sample.
Cell Purification and In Vitro Studies
To isolate unfractionated T cells, spleen cell suspensions were maintained in complete RPMI-1640 medium supplemented with 5% fetal calf serum (FCS). Cells were washed twice with Hank's balanced salt solution (HBSS) and incubated with Thy1.2 magnetic-activated cell separation (MACS) microbeads, respectively, in HBSS containing 5% FCS and 2 mM EDTA for 15 min at 4°C. After incubation, cells were positively selected on MS MACS separation columns (Miltenyi, Biotec, Auburn, CA). MDSCs from normal or Neupogen-treated mice were purified from spleen cell suspensions after incubating with PE-conjugated Gr-1 and anti-PE microbeads using a similar protocol as above. For carboxyfluorescein succinimidyl ester (CFSE) dilution assays, purified T cells were labeled with CFSE at 2.5 μM concentration and incubated at 37°C for 10 min. Cells (1 × 106) were cultured with 1 × 106 antigen-presenting cells (APCs) (T-cell depleted, mitomycin-treated splenocytes) in the presence or absence of 50 μl anti-CD3 supernatant (2C11) in 24-well plates for 48 h. MDSCs were then added at 1:1 ratio to responding T cells. In other experiments, purified T cells were isolated from naive BALB/c mice, or mice treated with Neupogen and/or IL-2 complex and then incubated with C57BL/6 APCs (1 × 106) in the presence or absence of MDSCs (as indicated) obtained from Neupogen-treated mice. [3H]Thymidine was added to cultures in the last 18 h of a 96-h culture.
In Vivo Mouse Studies
Mice were injected daily with recombinant human G-CSF (Neupogen, Amgen Inc., Thousand Oaks, CA at 5 μg/mouse) subcutaneously (SC) once a day for 5–14 days unless otherwise specified. G-CSF was diluted in 5% dextrose and 1% mouse serum albumin (vehicle). As control, mice received vehicle or PBS. IL-2 complex (IL-2C) containing 0.5 μg recombinant mouse IL-2 (eBioscience, San Diego, CA) and 5 μg rat anti-mouse IL-2 (JES6–12A1, R&D Systems, MN) in 100 μl PBS was injected into the peritoneal cavity of each mouse for 5 consecutive days and twice a week thereafter (48) throughout the duration of the study for skin graft recipients. Skin grafting was performed as previously described (1). Grafts were monitored every other day and scored as rejected when >80% or more of the original graft tissue had become necrotic as assessed by visual examination.
Histological Assessment
Donor skin tissues were harvested from recipient mice at indicated time points posttransplant and fixed in paraformaldehyde. Tissue sections (5 μm thick) were then stained with hematoxylin-eosin after paraffin embedding.
Statistical Analysis
Data are expressed as mean ± SEM unless otherwise indicated. A two-tailed Student's t-test was used to determine the significance between two groups as specified in the results. Values of p < 0.05 were considered significant. Bonferroni correction test was performed for all compared groups at indicated confidence levels to further evaluate the validity of the significance in differences as measured by the Student's t-test.
Results
In Vivo Administration of Human Recombinant G-CSF (Neupogen) Induces MDSCs in Mice
To begin to investigate whether these suppressor cells are efficacious in promoting tolerance to alloantigens, we treated mice with recombinant human G-CSF (Neupogen), the administration of which has been reported to increase the fraction of cells that express Gr-1, one of the phenotypic markers of MDSCs (37). A short course (5 days) of treatment with different doses of Neupogen led to induction of Gr-1+CD11b+ cells in the spleen of BALB/c recipient mice, which was remarkable at the 5 and 10 μg dosage 1 day posttreatment (Fig. 1A-C). This effect was transient, as only modest increase above control levels in the MDSC frequency was noted at 3 and 7 days after treatment (Fig. 1A, C). From these data, we determined that a dosage of 5 μg/recipient was optimal at inducing MDSCs and was chosen for subsequent treatments. To further evaluate the efficacy of in vivo Neupogen administration, we analyzed, concurrently, multiple tissues including peripheral blood, spleen, bone marrow (Fig. 1D), and lymph nodes (data not shown). Similar to the spleen, significant increases in the fraction of Gr-1+CD11b+ MDSCs were observed. As this induction appears to be short lived, a 2-week daily injection regimen was then utilized to determine whether chronic treatment affects the duration of MDSCs induction and persistence. Upon analysis, elevated levels of MDSCs could be observed in the spleen of treated mice for more than 1 week after cessation of the 14-day administration regimen (Fig. 1E). These data indicate that Gr-1+CD11b+ cells are effectively induced in mice using recombinant human G-CSF and highlight how chronic administration can enhance MDSCs persistence.

Kinetics of myeloid-derived suppressor cell (MDSC) induction in Neupogen-treated mice. BALB/c mice were injected SC with recombinant human granulocyte-colony stimulating factor (G-CSF; Neupogen) at 1, 5, and 10 μg (A) or 5 μg/mouse (B-E) for 5 (A-D) or 14 (E) consecutive days. Control mice received vehicle or PBS. At different time points as indicated in the graphs, mice were sacrificed and flow cytometry analysis was then performed to evaluate the frequency of MDSCs, identified by coexpression of CD11b and Gr-1. (A) Frequency of MDSCs in the spleen of mice treated with the indicated doses of Neupogen 1, 3, and 7 days after the last injection. (B) Representative fluorescence-activated cell sorting (FACS) plot 1 day posttreatment in the spleens of mice that received 5 μg of Neupogen (lower dot plot) or of control mice that received PBS (upper dot plot). (C) Absolute numbers of Gr-1+CD11b+ cells in the spleen of control versus Neupogen-treated mice at 1, 3, and 7 days posttreatment. (D) Summary of MDSC frequency in the peripheral blood (PB), bone marrow (BM), and the spleen (SPLN) of treated mice 1, 3, and 7 days after the last treatment. (E) Summary of MDSC frequency 1–2 weeks after the last injection following chronic (14 consecutive days) administration of Neupogen. Data represent the mean ± SEM for 2–5 mice/time point.
The Phenotype of Neupogen-Induced MDSCs Is Largely Indistinguishable From That in Naive Mice
In this study, it remains a possibility that MDCSs induced with Neupogen exhibit a distinct phenotype that is different from that seen in naive mice. To investigate this issue, MDSCs isolated from the spleen of Neupogen-treated mice were analyzed. We show that they express little to no MHC class II molecules, B7.1 (CD80) costimulatory molecule, and moderate level of F4/80, similar to MDSCs from tumor-free normal mice (Fig. 2A). However, a marginal increase was noted for CD40 expression on induced MDSCs relative to their naive counterparts. In addition, IL-4Rα, which has been correlated with suppressive function (16,42), was higher on MDSCs from Neupogen-treated mice than that on MDSC from naive mice (Fig. 2A, B, Table 1), suggesting that Neupogen not only expanded MDSCs but also induced their activation. We also found that expression of the CXCR2 and CCR4 chemokine receptors was much higher on MDSC from treated mice, consistent with the idea of systemic recruitment and trafficking that result in higher frequencies at multiple peripheral sites (data not shown). These data suggest that Neupogen administration not only increases MDSCs number but could also favor their maturation towards a more suppressive phenotype.
Summary of Mean Fluorescent Intensity (MFI) Values for the Expression of Indicated Cell Surface Molecules on Naive Versus Induced MDSCs as Described in Figure 2A

Values are MFI ± SEM.
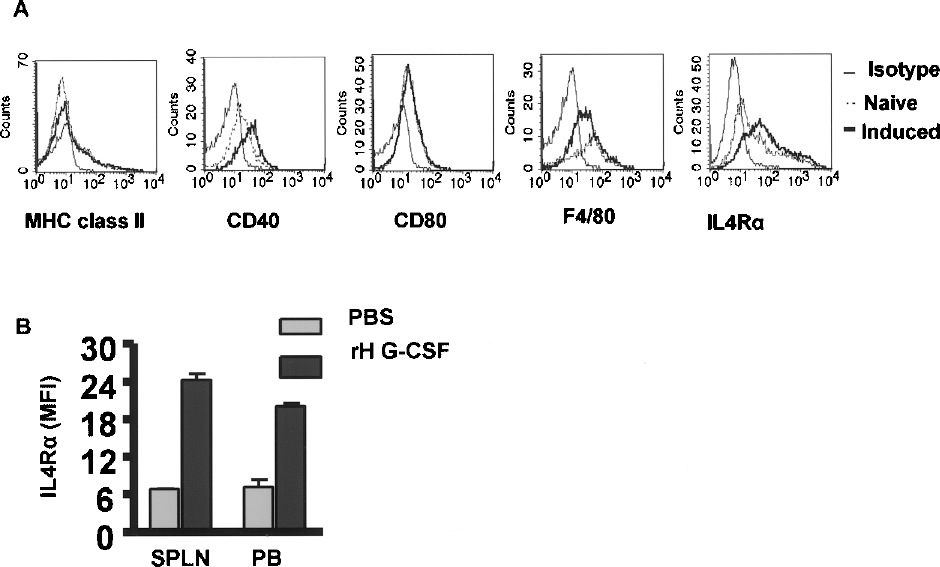
Phenotype of MDSCs in mice treated with Neupogen. BALB/c mice were treated with 5 μg Neupogen (recombinant human G-CSF) for 5 days. Multicolor FACS analysis was performed to establish the phenotype of induced MDSCs after gating on Gr-1+CD11b+ cells in the spleens of treated mice harvested 3 days after treatment. (A). Representative histogram plots of major histocompatibility complex (MHC) class II, CD40, CD80, F480, and IL-4Rα expression on gated Gr-1+CD11b+ cells in the spleen of naive (dashed lines) or Neupogen-treated mice (solid thick lines) analyzed 3 days after treatment. Isotype staining is depicted in solid thin line. (B) Mean fluorescent intensity (MFI) of IL-4Rα expression on gated MDSCs in the spleen (SPLN) and peripheral blood (PB) of naive versus Neupogen-treated BALB/c mice. Data are representative from three independent experiments.
Inhibition of T-Cell Response by Neupogen-Induced MDSCs
We then sought to determine whether MDSCs from Neupogen-treated mice are functionally capable of suppressing polyclonal and alloreactive T-cell responses. Unfractionated T cells stimulated with anti-CD3 proliferate extensively as revealed by CFSE dilution of both the CD4+ and CD8+ subsets within 3 days of culture (Fig. 3A). This proliferative response is inhibited upon addition of MDSCs isolated from Neupogen-treated mice with many responder cells (~38%) having undergone only one or two rounds of cell division (Fig. 3A). The addition of spleen cells, in contrast, failed to yield similar outcomes, demonstrating that the MDSCs suppression is unlikely due to nonspecific depletion of growth factors in the cultures. To further evaluate MDSC suppressive function, we set up a mixed leukocytes reaction (MLR) in the presence or absence of MDSCs. Analysis of T-cell cultures stimulated with allogeneic APCs revealed a significant reduction (p = 0.001) in proliferation of responder cells in the presence of MDSCs, while addition of control syngeneic spleen cells resulted in higher proliferative responses (Fig. 3B). Interestingly, on a per cell basis, MDSCs isolated from treated mice inhibited the MLR response more potently than those derived from naive unmanipulated mice (Fig. 3C). This finding supports our hypothesis that Neupogen promotes MDSCs differentiation toward a more suppressive phenotype. Collectively, these data demonstrate the suppressive capacity of Neupogen-induced MDSCs both in a polyclonal antigen stimulation setting and in an alloreactive response, which is highly relevant in transplantation settings.

Suppression of T-cell proliferative response by Neupogen-induced MDSCs. (A) BALB/c carboxyfluorescein succinimidyl ester (CFSE)-labeled T cells (1 × 106) were cultured with BALB/c antigen-presenting cells (APCs; 1 × 106) and stimulated with 0.5 μg (X-CD3 in 24-well plates for 3 days. MDSCs isolated from Neupogen-treated mice or spleen cells from normal mice were added to the cultures at a 1:1 ratio to the responding T cells. Proliferation of labeled T cells was then assessed by evaluating CFSE dilution as measured by FACS analysis after gating on viable 7AAD-, CD4+, or CD8+ T cells. Representative FACS plots of CFSE intensity on day 0 and day 3 of cell culture from three independent experiments are shown. (B) MDSCs from treated mice suppress alloreactive T-cell response in vitro. BALB/c T cells (1 × 105) were cocultured with C57BL/6 APCs (1 × 105) in 96-well round bottom plates for 4 days in the presence or absence of MDSCs (1 × 105) purified from the spleen of Neupogen-treated BALB/c mice or control spleen cells from normal mice. Thymidine was added to the triplicate wells in the last 18 h of culture. p-Values for statistically significant difference between the indicated pairs of experimental groups as determined by Student's t-test are shown (p < 0.05) and these differences remained statistically significant after performing Bonferroni correction test for comparisons to the no MDSC group at a 0.025 significance level. (C) Percent inhibition by induced versus naive MDSCs. Purified MDSCs isolated from naive or Neupogen-treated mice were added to cultures as described in (B) at indicated numbers. Percent inhibition was calculated from 3H proliferation counts in the presence of MDSC as a percentage of total counts in the absence of MDSCs. Data from two independent experiments are shown.
In Vivo Induction of MDSCs and Treg Cells Promotes Survival of Skin Allografts
Currently, there is paucity of data addressing MDSC-suppressive functions in transplantation settings. Because both Treg and MDSCs have the potential to prevent or attenuate alloreactive T-cell responses (1,10,12, 18,20,26,31) (Fig. 3B, C), we hypothesized that increasing their numbers in vivo by administration of Neupogen (for MDSCs) or IL-2C (for Treg cells) (48), alone or in combination might downregulate antidonor immunity and tip the balance in favor of tolerance. To test this hypothesis, we performed skin transplantation experiments with mice treated with Neupogen and/or IL-2C utilizing donor skin that is disparate in only the MHC class II constituents (i.e., bm12 to C57BL/6) as well as unrelated third party that is both MHC class I and II disparate (i.e., C3H to C57BL/6). In C57BL/6 recipients, treatment with Neupogen (Fig. 4B) resulted in a significant delay of MHC class II disparate (bm12) but not C3H allogenic donor skin rejection [p = 0.0045, mean survival time (MST) of 40 vs. 16 days] when compared to the control PBS-treated group (Fig. 4A). Interestingly, the administration of the IL-2 complex also delayed significantly (p = 0.002) the loss of bm12 donor skin (MST of 50 vs. 20 days for controls) (Fig. 4C), an effect that was even more pronounced when it was combined with Neupogen (MST of 74) (p = 0.0032) (Fig. 4D). Although the rejection of fully allogeneic C3H donor skin was comparable in all groups, these data highlight an effect of the combinatorial treatment in promoting prolonged survival across limited MHC barriers when the two naturally occurring suppressor cell subsets are induced to elevated levels in the periphery.

Kinetics of skin graft rejection in an MHC class II disparate skin graft model. Eightto12-week-old C57BL/6 mice received three contiguous skin grafts from C57BL/6, B6bm12, and C3H donor mice, respectively. Mice were then treated with Neupogen (5 μg) and/or IL-2 complex daily for the first 5 days beginning at day 0 and thereafter three times a week for Neupogen or twice a week for IL-2 complex throughout the duration of experiment. The control group received PBS. All recipient mice were monitored every other day until graft rejection, defined as >80% loss of tissue. Kinetics of graft rejection for MHC class II disparate bm12 donor skin and full MHC mismatch C3H donor skin in control C57BL/6 or treated recipients is shown. Treatments and the mean survival time (MST) of allogeneic skin grafts are listed within each panel of the figure.
Delayed Allograft Rejection Correlates with Attenuated T-Cell Response in Mice Treated with the Combination of Neupogen and IL-2 Complex
At the time of graft rejection, all transplant recipients were further analyzed to determine frequencies of MDSCs and Treg cells. Although previous findings have demonstrated that MDSCs can induce Treg expansion (19,27,44), treatment with Neupogen alone did not significantly increase Treg fractions in the peripheral blood, spleen, and lymph nodes (Fig. 5A). Administration of IL-2C alone or in combination with Neupogen, however, led to expansion of CD4+Foxp3+ Treg cells in these peripheral sites when compared to PBS controls (Fig. 5A). While Neupogen alone resulted in an elevation of MDSC fraction in treated mice, the percentage of MDSCs was not affected in mice treated with IL-2 complex, suggesting that the latter acts specifically to promote the expansion of Treg cells and not MDSCs (Fig. 5B). Interestingly, an additive effect was noted for MDSC fractions in mice treated with a combination of both IL-2 complex and Neupogen in all three compartments (Fig. 5B). One possible effect of the increased frequency of both of these naturally occurring suppressor cells in circulation is that T-cell response is dampened in treated mice. To evaluate T-cell response to alloantigens in vitro, T cells purified from C57BL/6 mice that were treated with Neupogen and/or IL-2C were stimulated in vitro with allogeneic APCs in the presence or absence of α-CD3. Upon analysis, the proliferative response of T cells derived from all groups as indicated after polyclonal stimulation was comparable (Fig. 5C). In MLR responses, however, proliferation by T cells obtained from mice treated with both Neupogen and IL-2 complex was significantly reduced in contrast to that of T cells obtained from mice treated with PBS, Neupogen, or IL-2 complex alone (Fig. 5D). These data suggests that alloantigen-reactive T-cell subsets in mice treated with both agents remained in a somewhat suppressed state ex vivo and thus only partially responded to alloantigen stimulation.

Induction of MDSCs and CD4+Foxp3+ Treg cells upon coadministration of Neupogen and IL-2 complex. C57BL/6 skin transplant recipients as described in Figure 4 were sacrificed at time of graft rejection and the peripheral blood, spleen, and lymph nodes (LN) were utilized for multicolor FACS analysis to determine the fraction of CD4+Foxp3+ or Gr-1+CD11b+ cells. Summary of (A) CD4+Foxp3+ Treg or (B) Gr-1+CD11b+ MDSC percentages in the peripheral blood (PB), spleen (SPLN), and lymph nodes (LN) in the groups that received the indicated treatments. (C, D) C57BL/6 mice were treated for 5 consecutive days with Neupogen and/or IL-2 complex, followed by a maintenance dose of Neupogen (three times) and/or IL-2 complex (twice) for 1 week. Three days after last treatment, mice were sacrificed and T cells (1 × 105) purified from the spleens of these mice were cultured with BALB/c (left) or C3H (right) APCs in the presence (C) or absence (D) of (X-CD3 for 3 or 4 days, respectively, in 96-well round bottom plates. Thymidine was added to the triplicate wells in the last 18 h of culture. Summary of T-cell proliferation based on thymidine incorporation from one representative of two independent experiments is shown. p-Values for statistically significant difference between the indicated pairs of experimental groups as determined by Student's t-test are shown (p < 0.05) and the difference between the IL-2C and IL-2C + Neu in mice cultured with C3H APCs remained statistically significant (and other values were nearly significant) after performing Bonferroni correction test for comparisons to the IL-2C +Neu group at a 0.0167 significance level. (E, F) Evaluation of the CD3-ζ chain in treated mice. Mice were treated with Neupogen or IL-2 complex (IL-2C) alone or in combination for 5 days. Multicolor FACS analysis was performed to establish the expression of the CD3-ζ chain on T cells in the spleens of treated mice harvested 2 days after treatment. (E) Representative histogram plots and (F) summary of mean fluorescent intensity (MFI) of ζ chain expression on gated CD4+ (left panel) and CD8+ (right panel) cells in the spleen of indicated groups of treated mice. Data in (F) are a composite of 3–4 mice per group.
One of several mechanisms that is implicated in MDSC-mediated suppression of antitumor immunity is the downregulation of the ζ chain of the CD3 complex that results in diminished activation of tumor-specific T cells (15). Due to the prevalence of high fractions of MDSCs in mice treated with Neupogen, we investigated whether this mechanism is operative in our system. After a short course of Neupogen and/or IL-2 complex treatment, analysis of the splenic T cells revealed a downregulation of the ζ chain in Neupogen or IL-2 complex-treated mice that was more striking with the combination of the two agents (Fig, 5E, F). This result is consistent with the diminished alloreactive responses by T cells from treated mice and supports the notion that impaired T-cell activation resulting from suboptimal CD3 signaling likely contributes to lowered T-cell proliferation.
Histological Assessment of Skin Grafts Reveals Markedly Reduced Cellular Infiltration in Treated Recipients
In addition to visual evaluation and scoring of donor skin grafts, we performed hematoxylin and eosin staining (H&E) on donor bm12 skin tissues that were excised from C57BL/6 transplant recipients treated with PBS (controls), Neupogen, and/or IL-2C. Analysis at day 12 posttransplant revealed that in the control PBS group there was massive cellular infiltration and tissue damage, which precedes graft loss between 15 and 20 days for rejecting control mice. In contrast, intact epidermis, hair follicles, and minimal infiltrates were observed in all experimental mice (Fig. 6A). Remarkably, at 60 days posttransplant, 2/4 of the mice treated with IL-2 complex and 4/5 of the mice treated with Neupogen/IL-2 complex still retained 50–70% of donor graft (data not shown). Histological evaluation of bm12 graft retrieved from these mice at day 60 posttransplant revealed moderate infiltration, but with a mostly intact epidermal layer and preserved hair follicles, suggesting minimal pathological damage to the overall tissue (Fig. 6B).
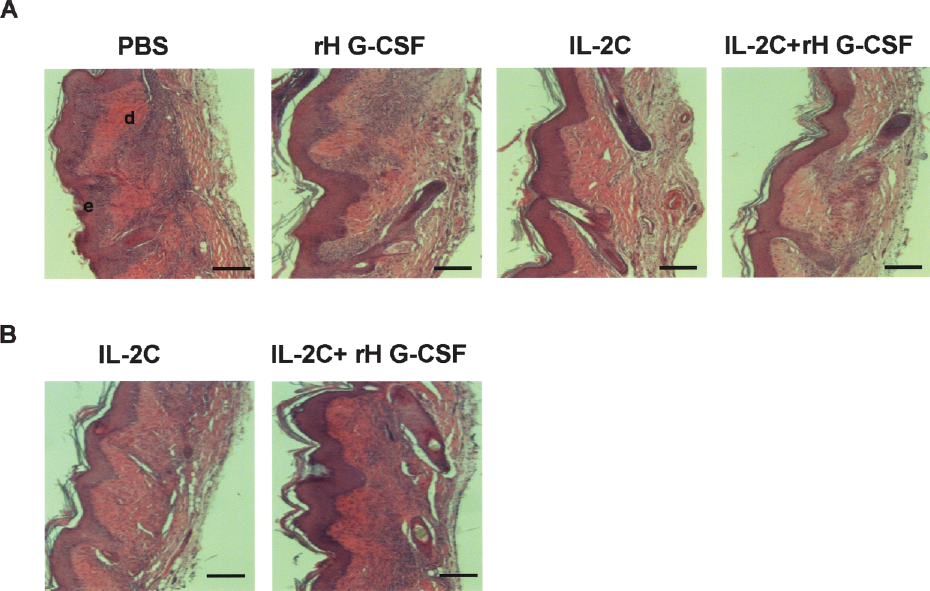
Histological evaluation of skin grafts. Donor C57BL/6 bm12 skin were harvested at (A) 12 days from C57BL/6 mice that were treated as indicated or at (B) 60 days for the IL-2 complex or in combination with Neupogen-treated group only. Tissue sections (5 μm thick) were stained with hematoxylin and eosin (H&E) after paraffin embedding. Scale bars: 125 μm. e; epidermis, d; dermis.
Taken together, these data further support the overall observation that loss of bm12 donor skin is delayed in treated mice, possibly due to inhibition of effector cell infiltration into the graft or impairment of effector function at the graft site.
Discussion
Current treatments for restraining the immune responses against allografts often utilize strong immunosuppressive biological agents that can sometimes lead to damaging side effects and generate a global depression of the immune system. As various regulatory and/or suppressor cells that target multiple immune perturbations are being uncovered, there is increasing interest in the utilization of such naturally occurring suppressive cells as therapeutic alternatives to promoting transplantation tolerance. Herein, we explored the potential of two suppressive leukocyte subsets, CD4+Foxp3+ T cells and myeloid-derived suppressor cells, to facilitate tolerance in a murine model of MHC disparate skin transplantation. A key finding in this study is that donor skin graft rejection is significantly delayed when the fraction of these two regulatory cells are increased in circulation as potentiated by IL-2 complex and recombinant human G-CSF.
In many experimental tumor models, a high fraction of MDSCs are observed in the tumor microenvironment and this correlates with an abundance of immunomodulatory cytokines such as G-CSF, GM-CSF, and IL-10, which are secreted by tumor cells demonstrating the role of these biological agents in the recruitment of MDSCs (15,29). Our finding that administration of recombinant human G-CSF (Neupogen) led to several fold induction of MDSCs in multiple tissues supports such observations and is consistent with reports of others where GM-CSF and/or G-CSF have been utilized to mobilize cells of the myeloid lineage in rodents (28,37,43). Although significant large numbers of MDSCs relative to unmanipulated animals were measured in the peripheral blood, spleen, and bone marrow, the fold increase is less dramatic and somewhat unremarkable in the lymph nodes. As a critical site for T-cell priming, lymph node MDSCs may be instrumental in creating a “tolerogenic” environment. Thus, treatments targeted at induction or trafficking of MDSCs to lymph nodes need to be explored. Of note is that our choice of human G-CSF for these experiments is in part based on our findings that both mouse and human G-CSF were superior in inducing MDSCs in vivo compared to GM-CSF (data not shown). We chose human G-CSF for these experiments due to its ready availability and its potential use in additional investigations that may utilize human cells in a murine model. Although the dose of the human G-CSF utilized in this study is higher than currently administered in patients after chemotherapy, it is important to reiterate that our investigations were performed in mice. So, the observation that mouse G-CSF induced levels of MDSCs that were higher than human G-CSF in one of our limited experiments (not discussed) is not surprising. Thus, we have reason to believe that current clinical dosage of Neupogen or even lower may be efficacious for promoting the induction of MDSCs in humans due to specie specificity. Moreover, because interindividual differences may exist in the metabolism and catabolism of this cytokine (32), further studies in humans are necessary to establish the minimal dose of Neupogen capable of mobilizing and differentiating MDSCs for therapeutic purposes.
An outstanding issue is whether the induced MDSCs are different in phenotype and function from the naturally occurring cells. Given the heterogenicity of MDSCs, we cannot rule out the possibility that other minor immature myeloid progenitor cellular subsets reside within the induced population. Nevertheless, it is quite clear from our assessments that the induced MDSCs are largely similar in phenotype to the typical Gr-1+CD11b+ cells in naive mice. The higher expression of IL-4Rα expression on the former, however, may indicate a more potent suppressive function, consistent with the observation that MDSCs from Neupogen-treated mice inhibited T-cell proliferation better than those derived from naive mice when tested in an MLR (Fig. 3C). Our evaluation of the induced MDSCs in the present study also revealed they express little to no MHC class II molecules and other canonical costimulatory molecules, and, as such, are unlikely to function and contribute to professional and classical antigen presentation and T-cell activation. Although we previously showed (44), using a transgenic system, that MDSCs can cross-present antigen specifically to Tregs, it is extremely difficult to confirm or deny this hypothesis in this experimental setting because skin alloantigens are unknown and CD4 T cells with this particular specificity are not easily detectable.
Our data indicate that both CD4+ and CD8+ T cells were effectively suppressed in the presence of polyclonal anti-CD3 stimulation, consistent with the report by De Wilde et al. (10). The capacity of these MDSCs to prevent alloantigen-mediated T-cell response was demonstrated in an MLR and further extended to in vivo studies where MHC class II disparate bm12 donor skin graft rejection was delayed significantly in the presence of induced MDSCs. In naive mice, the kinetics of allogeneic skin graft rejection is not altered even in the presence of a small fraction of circulating MDSCs, suggesting that utilization of these cells to favor transplantation tolerance will likely require protocols geared at expanding their numbers in vivo. Our findings thus provide some proof of principle to achieving such goals, at least in a somewhat stringent animal model of MHC-mismatched transplantation. Interestingly, combinatorial treatment that also increases the frequency of CD4+Foxp3+ Treg cells in parallel with MDSCs strikingly led to more robust delay of MHC class II disparate donor skin, suggesting an additive suppressive effect on host alloreactive T cells. Consistent with this idea, T cells isolated from mice treated with both IL-2 complex and Neupogen proliferate less in the presence of allogeneic APCs when compared to those derived from mice receiving single treatments (Fig. 5D). Although this in vitro finding suggests some attenuation of alloreactive T-cell response that, for instance, correlates with the ex vivo analysis of CD3 ζ chain expression on T cells isolated from treated mice, it is somewhat contradictory to the in vivo observations in which C3H donor skin graft was not delayed in mice treated with both Neupogen and IL-2 complex. Our in vitro study represents an isolated event involving alloreactive T-cell response, particularly, through the direct allorecognition pathway. In vivo, however, multiple effector mechanisms have been implicated in acute and/or chronic graft rejection. Besides the indirect pathway, which is also crucial for chronic graft rejection, there is growing evidence that some humoral (high-affinity alloantibodies) and even innate (NK cells) immune response contribute to graft rejection (22,30). Thus, the apparent disparity between depressed alloreactive T-cell response in vitro and the absence of a delay in fully allogeneic donor graft may likely be explained by such additional effector mechanisms that regulatory cells need to overcome to promote tolerance.
It is unclear at present whether this conditioning regimen results in a generalized immunosuppression similar to drugs that are currently utilized to dampen the immune response in transplantation settings or if it is a graft specific anergy. Our findings demonstrate that these treatment regimen are unlikely to induce a “wholesale” anergy as proliferation to polyclonal stimulator was only minimally diminished in vitro within the bulk T-cell population. Although the mechanisms of MDSC suppression in this study are yet to be elucidated, it remains a possibility that the induced MDSCs may modify T-cell phenotype, using mechanisms dependent on L-arginine metabolism and/or disrupting the CD3 complex (14,15,35,39,45,47,49), which ultimately results in lowered effector function. The finding that the ζ chain expression by T cells within the spleen of mice treated with either Neupogen or more remarkably in combination with IL-2 complex is downregulated is consistent with this notion. It remains to be determined the possible involvement of Treg cells in this process as IL-2 complex treatment, similar to Neupogen treatment alone, led to lower expression of the ζ chain. Alternatively, MDSCs may induce the apoptosis of some of the alloreactive T cells that are in proximity within a microenvironment (15) and this may explain the significantly depressed proliferative response by T cells from treated mice. Another possibility is that when present in higher than normal frequencies within a niche, they may competitively prevent APCs from gaining sufficient access to T cells, thus increasing the threshold required for optimal T-cell activation and differentiation. Although we favor the idea that our approach utilizing pharmacologic agents to induce MDSCs and Tregs in vivo may be less harsh on the immune system than commonly used immunosuppressive drugs, additional experiments are currently under way to evaluate these possibilities.
Accumulation of suppressive T cells has been previously noted in long-term tolerated grafts (17). Histological assessment revealed minimal infiltration of inflammatory cells within donor skin from IL-2 complex/Neupogen-treated recipients when retrieved during the acute phase of graft rejection, but over time (day 60) some inflammation became evident. One possible explanation for our histological findings is that T-cell differentiation is initially effectively prevented in draining lymph nodes, but with time, chronic graft rejection coincides with activated T cells escaping complete suppression and migrating to grafts. The near absence of severe damage and necrosis of “tolerated” grafts even in the presence of moderate level of leukocytic infiltrates may be an indication that the induced suppressive cells also migrate to the graft to actively curb host–antidonor immune response. Additional experiments are planned to investigate these issues.
To date, only a few reports have explored the possible involvement of MDSCs in transplantation tolerance (10,12,31,53). Our investigation differs from that of others in that recipient mice did not undergo extensive conditioning or were not subjected to cellular ablation protocols (5) that deplete potential effector cells. We relied on the potential for naturally occurring suppressor cells to inhibit “allo” immunity by utilizing Neupogen, a drug that is widely used in the clinic to promote myeloid cell survival, expansion, and differentiation, as well as IL-2 complex, which effectively boosts the expansion of Treg cells, similar to IL-2 therapy (2,52) or superagonist CD28 antibody treatment (21). While these observations indicate that both MDCSs and Treg cells can “cooperatively” promote transplantation tolerance, it is of interest to investigate ways to expand these cells in an antigen-specific context as this provides the benefit of tolerance to MHC antigens while allowing immune response to foreign pathogens.
In summary, our findings highlight the potency of MDSCs in combination with Treg cells to inhibit allograft rejection and may provide therapeutic strategies to induction and maintenance of transplantation tolerance. In addition, our model provides a platform to investigate tolerance to vascularized solid tissues in broader transplantation experiments as well as to evaluate mechanisms by which both cells synergistically promote tolerance.
Footnotes
Acknowledgment
This work was supported by the Diabetes Research Institute Foundation.