
Abstract
Mesenchymal stromal cells (MSCs) are attractive for cellular therapy of muscular dystrophies as they are easy to procure, can be greatly expanded ex vivo, and contribute to skeletal muscle repair in vivo. However, detailed information about the contribution of bone marrow (BM)-derived human MSCs (BM-hMSCs) to skeletal muscle regeneration in vivo is very limited. Here, we present the results of a comprehensive study of the fate of LacZ-tagged BM-hMSCs following implantation in cardiotoxin (CTX)-injured tibialis anterior muscles (TAMs) of immunodeficient mice. β-Galactosidase-positive (β-gal+) human-mouse hybrid myofibers (HMs) were counted in serial cross sections over the full length of the treated TAMs of groups of mice at monthly intervals. The number of human cells was estimated using chemiluminescence assays. While the number of human cells declined gradually to about 10% of the injected cells at 60 days after transplantation, the number of HMs increased from day 10 onwards, reaching 104 ± 39.1 per TAM at 4 months postinjection. β-gal+ cells and HMs were distributed over the entire muscle, indicating migration of the former from the central injection site to the ends of the TAMs. The identification of HMs that stained positive for human spectrin suggests myogenic reprogramming of hMSC nuclei. In summary, our findings reveal that BM-hMSCs continue to participate in the regeneration/remodeling of CTX-injured TAMs, resulting in ±5% HMs at 4 months after damage induction. Moreover, donor-derived cells were shown to express genetic information, both endogenous and transgenic, in recipient myofibers.
Keywords
Introduction
Skeletal muscle tissue contains multinucleated myofibers that arise from fusion of mononucleated progenitor cells (i.e., myoblasts). The ability of skeletal muscle tissue to regenerate during the life time of an individual is dependent upon a population of resident stem cells, designated satellite cells. In response to skeletal muscle injury, the normally quiescent satellite cells get activated, replicate, and differentiate into myoblasts. These in turn are incorporated into damaged myofibers or fuse among themselves to form new myosyncytia.
Duchenne muscular dystrophy (DMD) is characterized by irreversible skeletal muscle damage due to mutations in the sarcolemma-stabilizing dystrophin protein. Exhaustion of the satellite cell reservoir eventually leads to excessive muscle degeneration. Cell-based therapy has gained much attention as a strategy to provide dystrophin-deficient myofibers with functional copies of the DMD gene and thus to interfere with disease progression. This approach entails transplantation of either allogenic cells from a healthy donor (43,55,58) or autologous cells genetically modified with sequences that can either complement or correct the gene defect (2,29,49). Its success relies on the incorporation of these cells into regenerating myofibers. Progenitor cells derived from both skeletal muscle (34,49,55,57) as well as from other tissues (1,13, 21,52,59) have been explored for this purpose (6,44,47). Although experimental and clinical studies with myoblasts have yielded important insights, their clinical application is hampered by (i) limited availability, (ii) restricted ex vivo proliferation capacity, and (iii) limited spread in host muscles (45,56,60).
Mesenchymal stromal cells (MSCs) may represent an attractive alternative for myoregenerative purposes. MSCs, first isolated from the bone marrow (BM) (22), are adherent fibroblast-like cells that can differentiate to adipocytes, chondrocytes, and osteoblasts in vitro (3,16). Their isolation relies on adhesion to cell culture plastics. In culture, human MSCs (hMSCs) are capable of extensive proliferation without showing chromosomal aberrations (48).
Because antigens exclusively displayed on the plasma membrane of MSCs have not been identified yet, these cells are phenotypically characterized by a combination of several nonhematopoietic cell surface markers (9,10). Cells meeting these characteristics have been isolated from a variety of human tissues, including adipose tissue (AT) (66), synovial membrane (SM) (11), skeletal muscle (64), the circulatory system (38), dental pulp (27), skin (28), menstrual blood (46), umbilical cord (20), amniotic fluid (32), placenta (31), fetal blood, spleen, lungs, liver, and BM (30,65).
Recent studies have shown that hMSCs from different tissues acquire properties of skeletal muscle cells when exposed to myogenesis-promoting culture conditions (11,14,63) or in response to forced synthesis of myogenic transcription factors (25,26). Furthermore, in cocultures with myoblasts, hMSCs can engage in myotube formation through heterotypic cell fusion (23,40, 54). Due to their high transducibility by viral vectors (8,36), hMSCs can also be used as delivery vehicles of recombinant genes into dystrophin-deficient myotubes (23,25,40,54).
In vivo, both murine (15) and human MSCs have been shown to contribute to the regeneration of dystrophic and experimentally damaged skeletal muscles. In the case of the hMSCs this capacity has been demonstrated for cells derived from bone marrow (BM-hMSCs) (14,54), adipose tissue (AT-hMSCs) (41,50), and synovial membrane (SM-hMSCs) (12). Most of these studies were concerned with the contribution of naive hMSCs to skeletal muscle regeneration. In this context, the term naive refers to cells that have been expanded in culture but were not modified by transduction or otherwise to acquire myogenic properties. Such naive cells seem to offer significant advantages for clinical use because safety precautions are less complicated. In fact, naive BM-derived hMSCs (BM-hMSCs) have already been used in several clinical studies for the treatment of refractory graft-versus-host disease following allogenic BM transplantation (39). In the context of skeletal muscle diseases, however, the available information on naive BM-hMSCs is limited to the demonstration of a few enhanced green fluorescent protein (eGFP)-positive fibers on day 7 after local injection of eGFP-marked hMSCs into cardiotoxin (CTX)-injured tibialis anterior muscles (TAMs) of immunodeficient (i.e., NOD/SCID β2m–/–) mice (54).
In this study, a detailed evaluation of the capacity of BM-hMSCs to contribute to skeletal muscle repair is presented. We employed CTX-induced TAM injury followed by local injection of LacZ-transduced hMSCs in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice to obtain a quantitative description of the participation of BM-hMSCs in the regeneration/remodeling process over a period of 120 days after injury.
Materials and Methods
Isolation and Culture of hMSCs
All experiments were performed with BM cells of a 38-year-old female collected with informed consent during orthopedic surgery according to the guidelines of the Leiden University Medical Center (LUMC, Leiden, the Netherlands). hMSCs were isolated from the BM sample as described previously (36). At passage number 2, aliquots of 2 × 105 cells were cryopreserved. hMSC expansion was performed in culture medium containing 0.5 ng/ml basic fibroblast growth factor (FGF2; Sigma-Aldrich, St. Louis, MO) unless specified otherwise. Cell doubling time was calculated as described previously (36) from cultures initiated by seeding 5 × 104/cm2 of freshly thawed cells in 25-cm2 cell culture flasks (Greiner bio-One, Frickenhousen, Germany). After reaching 70–80% confluence, the total number of cells in each culture was determined and a fraction of them was replated for a next passage under the same conditions as above. Cell replication was monitored for up to 15 passages.
The characterization of the cultured BM-hMSCs used in this study by immunophenotyping and in vitro differentiation assays has been reported previously (36,61).
Lentivirus Vector Production
The vesicular stomatitis virus G protein-pseudotyped self-inactivating human immunodeficiency virus type 1-based vector LV.C-EF1a.cyt-bGal, which codes for wild-type Escherichia coli β-gal, was produced in 293T cells using shuttle plasmid pLV.C-EF1a.cyt-bGal.dCpG and packaging constructs psPAX2 (Addgene, Cambridge, MA) and pLP/VSVG (Invitrogen) as previously described (62). Lentivirus vector particles were purified by filtration through 0.45-μm pore size cellulose acetate filters (Pall, Mijdrecht, the Netherlands) and centrifugation through 20% (w/v) sucrose (VWR International, Amsterdam, the Netherlands) cushions. The shuttle plasmid pLV.C-EF1a.cyt-bGal.dCpG contains a hybrid internal promoter consisting of the enhancer of the murine cytomegalovirus immediate-early gene, the core promoter of the human elongation factor 1α gene, and a synthetic intron coupled to a CpG motif-free version of the Escherichia coli LacZ gene. The source of these genetic elements was the 3.9-kb Klenow-blunted EcoRI-NheI fragment of pCpGvitro-neo-LacZ (Cayla-Invivo-Gen Europe, Toulouse, France). The complete nucleotide sequence of pLV.C-EF1a.cyt-bGal.dCpG has been deposited in GenBank under accession number GQ853402.
BM-hMSC Transduction
Twenty-four hours before transduction with the lentivirus vector LV.C-EF1a.cyt-bGal, BM-hMSCs of passage 4 were seeded at a concentration of 4 × 104 cells/cm2. The cells were then incubated with 2 HeLa cell-transducing units of the vector/hMSC in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS; both from Invitrogen-Gibco, Breda, the Netherlands) and 8 μg/ml hexadimethrine bromide (polybrene; Sigma-Aldrich) for 4 h at 37°C. Transduction efficiency was determined 14 days later by staining the cells with X-gal solution containing 2 mM 5-bromo-4-chloro-3-indolyl-β-D-galactosidase (X-gal; Sigma-Aldrich), as previously described (24). Typically, more than 90% of the LV.C-EF1a.cyt-bGal-transduced hMSCs (LacZ-hMSCs) were β-gal+ at this time point as well as after multiple ex vivo cell doublings. After transduction, the cells were passaged twice and cryopreserved until further use. Before the start of an in vivo experiment, aliquots of the LacZ-hMSCs were thawed again and expanded for up to 4 additional passages to obtain the required number of cells. This procedure allowed us to carry out all in vivo experiments with LacZ-MSCs from a single cell batch and ensured the use of cells that had undergone ≤8 passages in culture.
Animal Model and Cell Transplantation Procedure
The NOD/LtSz-scid/scid/J (NOD/SCID) mice used in this study were obtained from our breeding colony at the animal facilities of the LUMC. This colony was established with animals originally purchased from Jackson Laboratories (Bar Harbor, ME). Male and female mice aged 8–12 weeks were used. Animals were housed as previously described (37). All experiments were performed according to a study protocol approved by the animal ethics committee of the LUMC.
Muscle regeneration was studied in an artificial damage model consisting of murine TAMs injected with 20 μl of a 20 μM Naja mossambica mossambica CTX (Sigma-Aldrich) solution in phosphate-buffered saline (PBS). Twenty-four hours after CTX injection, 5 × 105 LacZ-hMSCs of passage number 8 were suspended in 25 μl of PBS and injected at a single site in the center of the TAM. In each experiment, TAMs injected with CTX only served as negative controls. Mice were sacrificed at 3, 10, 30, 60, 90, or 120 days after cell injection. The TAMs were excised and either cryopreserved or fixed in 4% formaldehyde (Mallinckrodt Baker, Phillipsburg, NJ). Cryopreservation of the tissues was performed either by snap-freezing in liquid nitrogen (for chemiluminescence assay) or, after mounting of the tissue on a base of cork with tragacanth (Sigma-Aldrich), by submersion in liquid nitrogen-cooled 2-methylbutane (VWR International; for immunohistology). Tissues were stored at −80°C.
Histochemical Staining of Paraffin-Embedded Tissue Sections and Counting of Myofibers with Human Cell Contribution
To facilitate penetration of the fixative and staining solution, TAMs were divided transversally into two equal parts and fixed in 4% formaldehyde for 1 h at room temperature (RT). After rinsing three times for 5 min with PBS, the tissues were incubated overnight at 37°C in X-gal staining solution. Next, the TAMs were subjected to three 5-min washes with PBS and fixed again with 4% formaldehyde for 1 h at RT. Thereafter, tissues were embedded in paraffin and cut into 6-μm-thick serial transversal cross sections. Five consecutive sections were mounted per slide, representing a muscle stretch of 30 μm. In total 200 slides per muscle were prepared. The sections were costained with hematoxylin, phloxin, and saffron (HPS) following standard procedures. Images were captured with a digital camera (Color View IIIu) mounted on an Olympus BH-2 microscope and processed with the aid of cellF software (Olympus, Zoeterwoude, the Netherlands).
To determine the total number of hybrid myofibers (HMs) in a TAM, all sections on every fourth slide were examined for the presence of β-gal+/blue myofibers allowing the analysis of 30-μm-long tissue stretches at 90 μm distance along the entire length of the muscle. The total number of β-gal+ myofibers within one TAM was thus derived from the analysis of 250 sections. For each slide, the average number of blue myofibers found in the five individual sections was calculated. This number was then compared to that of the next slide. When the average number of β-gal+ myofibers in a slide exceeded that of the previous one, the difference was regarded to reflect the formation of additional HMs. The average number of blue myofibers in the first slide plus the additional HMs found in consecutive slides was considered to represent the total number of β-gal+ myofibers per TAM. To facilitate comparison of our data with those of others presenting values derived from single sections only, we also specified for each TAM the maximal number of blue myofibers found in a single section.
Histochemical and Immunofluorescent Staining of Cryopreserved Tissue Sections
Cryopreserved TAMs were cut into 6-μm-thick consecutive transversal cross sections that were placed on SuperFrost Plus glass slides (Menzel-Gläser, Braunschweig, Germany) and stored at −80°C.
X-gal staining was performed on several transversal cryosections along the whole TAMs after their fixation with 0.25% glutaraldehyde (grade II, Sigma-Aldrich) in PBS at 4°C for 15 min. After being air-dried and washed three times for 5 min with PBS, the sections were incubated overnight at 37°C in X-gal staining solution. The excess solution was removed by rinsing the slides thrice with PBS. The slides were fixed again with 0.25% glutaraldehyde at 4°C for 15 min. After washing the slides three times for 5 min each in PBS, the sections were counterstained with nuclear fast red and saffron following standard procedures. The slides were then mounted in Pertex mounting medium (Histolab Products, Gothenburg, Sweden). Images were captured as described above.
Several sections adjacent to sections containing β-gal+ myofibers were subjected to immunofluorescence staining with antibodies specific for human lamin A/C and human spectrin. These sections were fixed with acetone (Mallinckrodt Baker) for 10 min at −20°C, air-dried, and washed three times for 5 min with PBS. Next, the sections were incubated in a moist chamber with antibodies specific for human lamin A/C (Vector Laboratories, Burlingame, CA; clone 636; mouse IgG2b; 1:200; 1 h at RT) and human spectrin (Leica Microsystems-Novocastra Reagents, Valkenswaard, the Netherlands; clone RBC2/3D5; mouse IgG2b; 1:50; 6 h at 4°C) and subsequently with AlexaFluor 568-conjugated goat anti-mouse IgG2b (Invitrogen-Molecular Probes; 1:300; 1 h at RT). The primary antibodies were diluted in PBS-10% normal goat serum (NGS; Invitrogen-Gibco); the secondary antibody was diluted in PBS. After each incubation step, sections were rinsed three times for 5 min each in PBS. Next, the sections were stained for 10 min at RT with 1 μg/ml of Hoechst 33342 (Invitrogen-Molecular Probes) in PBS, washed thrice with PBS to remove excess dye, and mounted in Vectashield mounting medium (Vector Laboratories). Light microscopic analysis was performed with a Leica DM5500 B fluorescence microscope (Leica Microsystems, Rijswijk, the Netherlands). Images were captured with a CoolSNAP K4 CCD camera (Photometrics, Tuscon, AZ) and archived using home-made software.
Quantification of β-gal Activity in TAM Extracts
The human cell content in murine TAMs at different time points after intramuscular transplantation of LacZ-hMSCs was estimated using the Beta Glo assay system (Promega Corporation, Madison, WI). Immediately after surgical excision, TAMs were snap-frozen in liquid nitrogen and stored at −80°C until processing. Each whole TAM was homogenized in 1 ml of homogenization buffer (10 mM KCl, 1.5 mM MgCl2, and 10 mM Tris-HCl, pH 7.4) on ice as described by Eppens et al. (19). Where indicated, a cocktail of protease inhibitors (Complete, Mini, EDTA-free; Roche Diagnostic Nederland, Almere, the Netherlands) was present during the homogenization procedure. The homogenate was cleared by centrifugation for 10 min at 20,800 × g and 4°C. The supernatants were analyzed for β-gal activity according to the protocol provided by the manufacturer of the Beta Glo assay system. Chemoluminescence was measured using a Wallace 1420 VICTOR 3 multilabel plate reader (PerkinElmer Nederland, Groningen, the Netherlands). β-gal activity was expressed in relative light units (RLUs). Serial dilutions of each supernatant were analyzed and plotted against RLUs. RLUs of different samples were compared at a dilution positioned in the linear range of the dose–response curve.
Statistical Analysis
Data were expressed as the mean ± SD of each experimental group (30, 60, 90, and 120 days). Differences among groups were statistically analyzed using a one-way ANOVA test following by post hoc LSD test (α = 0.05). A value p < 0.05 was considered significant.
Results
The Effect of FGF2 on the Expansion and Biological Activity of BM-hMSCs
Cryopreserved cells of passage 2 were thawed and subsequently used to evaluate the effect of FGF2 on BM-hMSC proliferation in culture. As shown in Figure 1, all FGF2 concentrations tested enhanced BM-hMSC replication. The hMSCs cultured in medium without extra FGF2 underwent 18 population doublings in 138 days. The replication of BM-hMSCs cultured with FGF2 (all concentrations) leveled off around 120 days of culture reaching between 38 and 44 populations doublings in 131–148 days.
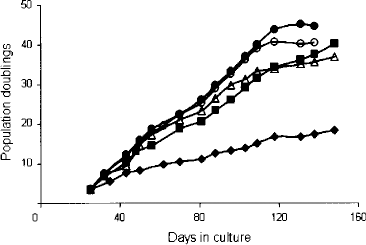
FGF2 enhances the replication of cultured BM-hMSCs. The graph shows a typical example of the effect of FGF2 on the replication rate of BM-hMSCs. Similar dose–response relationships were observed with BM-hMSC samples from other donors (data not shown). No FGF2 added (filled diamonds), cultures containing 0.5 ng/ml (filled squares), 1.0 ng/ml (open triangles), 5 ng/ml (filled circles), or 10 ng/ml (open circles) FGF2.
Based on these results BM-hMSCs were propagated in culture medium containing 0.5 ng/ml FGF2 for the remainder of the study.
Continuous Incorporation of hMSCs Into Regenerating Murine Myofibers
The contribution of BM-hMSCs to skeletal muscle regeneration was studied in CTX-injured TAMs of NOD/SCID mice. LacZ-hMSCs injected into the TAM 24 h after CTX administration were traced at various time points after transplantation by systematic screening of histological sections. The evaluation of transversal cross sections at regular intervals along the entire TAM allowed the detailed analysis of whole muscle morphology and β-gal signal distribution.
The injection of CTX in the center of the TAM caused destruction of almost the entire muscle (Fig. 2B). At 24 h and 3 days after treatment with CTX, the majority of the TAM was necrotic and disorganized, with infiltration of inflammatory cells. At 10 days post-CTX injection, numerous clusters of regenerating myofibers identified by their small diameter, basophilic cytoplasm, and centrally localized nuclei were present. After this acute phase of myoregeneration a period of muscle remodeling ensued. The latter period was characterized by the reorganization of myofibers in fascicles and enlargement of the diameters of the myofibers predominantly during the first month. Areas with myofibers of heterogeneous diameters were still present at 120 days. The central position of the nuclei in the vast majority (>90%) of the myofibers along the entire TAM at 4 months post-CTX injection illustrates the extensiveness of the repair process that took place.

Histochemical analysis of transversal sections of TAMs. (A) Untreated NOD/SCID mouse. (B) TAMs excised at the indicated time points after CTX injection (C) X-gal staining of cultured BM-hMSCs at 14 days after transduction with LV.C-EF1a.bGal. (D) TAMs collected at different time points after CTX treatment and implantation of LacZ-hMSCs. All sections were stained with X-gal and counterstained with HPS. Sections were selected for the presence of HMs, which frequently occurred in clusters. Although there is a leakage of the X-gal reaction product in all tissues (pale blue) the β-gal+ cells and myofibers are well defined by the dark blue color. The magnification of the micrographs in the left column is 100×. In the right column, fourfold magnifications of the areas delineated in the left column are shown.
The histological analysis did not reveal any obvious effect of the injection of LacZ-hMSCs on the healing of the CTX-damaged TAMs (compare Fig. 2B and D). Three days after hMSC administration, β-gal+ mononucleated cells were distributed throughout the necrotic tissue (Fig. 2D). Participation of human cells in the regeneration process became visible as early as 10 days after CTX injection by the appearance of few small β-gal+ myofibers. At 1 month after hMSC administration, β-gal+ myofibers were more abundant. The diameter of these myofibers differed greatly, indicating that they were formed at different times or that their growth rate was different.
In Figure 3, the number of β-gal+ myofibers in transverse cross sections along the length of the TAM is depicted for each individual mouse at different time points after administration of LacZ-hMSCs (Fig. 3A). In all instances, the number of HMs was higher in the central area of the TAMs than in their distal and proximal parts. The number of HMs showed a gradual increase over time, reaching an average of 104 (57–162) at 120 days post-CTX injection (Fig. 3B). Because murine TAMs contain approximately 2,000 myofibers (51 and our observation), at the end of the examination period donor cells had been incorporated in ±5% of the myofibers. At this time point, many of the β-gal+ myofibers were localized in clusters throughout the muscle (Fig. 2D).

Distribution and quantification of HMs (i.e., β-gal+ myofibers) in TAMs of NOD/SCID mice at 30, 60, 90, and 120 days after implantation of LacZ-hMSCs. (A) Average number of β-gal+ myofibers in transversal cross sections taken at regular intervals along individual TAMs. Each line represents one muscle. CTX and LacZ-hMSCs were administered in the center of the TAMs as indicated at the top. The interruption of lines in the middle of the muscle is due to lack of complete paraffin sections from initial cuttings. (B) Average number of HMs at 30 (n = 7), 60 (n = 7), 90 (n = 6), and 120 (n = 6) days after CTX injection. The black bars represent the average total number of β-gal+ myofibers per whole TAM (calculated as described in Materials and Methods). The white bars correspond to the average maximum number of β-gal+ myofibers in single cross sections of each TAM. Both values increase with time according to a linear regression (dashed lines). The slopes of the upper and lower dashed lines, which corresponds to the black and white bars, respectively, are 0.82 (R2 = 0.96) and 0.45 (R2 = 0.99). The average number of β-gal+ myofibers per whole muscle as well as the average maximum number of HMs per section at day 120 were statistically different (p < 0.05) from those at day 30 and 60 after CTX treatment.
At all time points analyzed, from day 10 after BM-hMSC injection on, single β-gal+ cells were identified in sections along a large part of the TAMs. In more than 50% (i.e., 17 of the 32) of the treated TAMs, human mononucleated cells were present from the proximal to distal end of the muscle (Fig. 4).
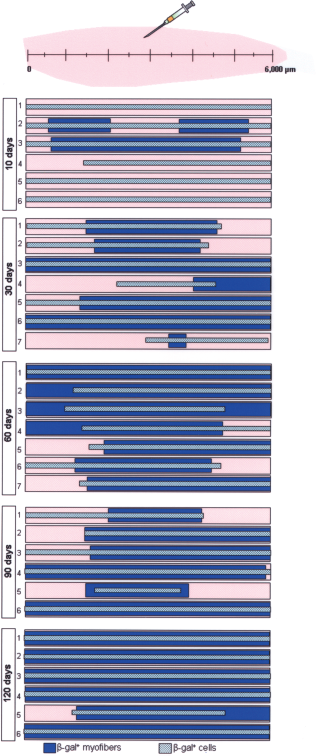
Distribution of β-gal+ cells and HMs along entire TAMs. The data result from the screening of X-gal-stained paraffin sections along entire TAMs as described in Materials and Methods. The length of the bars represents the parts of the muscle where β-gal+ cells (pale blue) or HMs (dark blue) were observed. Notice that β-gal+ cells are detected at the ends of the muscle as early as 10 days after their injection.
In all examined TAMs the X-gal reaction product in the myofibers was restricted to segments varying in length up to 2,000 μm. The analysis of series of adjacent sections often showed a bell-shaped distribution of the staining intensity with more intense blue in the central portion of the segments (Fig. 5A, B1, B2). Less frequently observed were short myofibers in which the X-gal reaction product was more or less evenly distributed along their entire length (Fig. 5B3).

Diffusion of β-gal in HMs. (A) β-gal activity in a single myofiber (arrows) as revealed by X-gal staining of transversal TAM sections spanning an area of 960 μm (a to f; the distance between presented sections is 160 μm). The sections were derived from a muscle collected at 1 month after injection of LacZ-hMSCs. The contribution of a human cell to this large myofiber is first visible in (a) by the light blue color. The staining intensity increases from (b) to (e). The presence in (f) of connective tissue in the area marked by the arrow indicates termination of the blue myofiber at a point between (e) and (f). (B) Representative longitudinal sections through TAMs containing HMs. In (B1) and (B2) myofibers with a short β-gal+ segment are displayed. In (B3) a short myofiber (arrow) is β-gal+ over its entire length. All samples were stained with X-gal and counterstained with HPS. Magnification 100× (A) and 200× (B).
Immunofluorescence staining of cryosections of TAMs excised 1 month after hMSC administration revealed the presence of human spectrin in β-gal+ myofibers (Fig. 6), indicating myogenic reprogramming of donor cell nuclei.
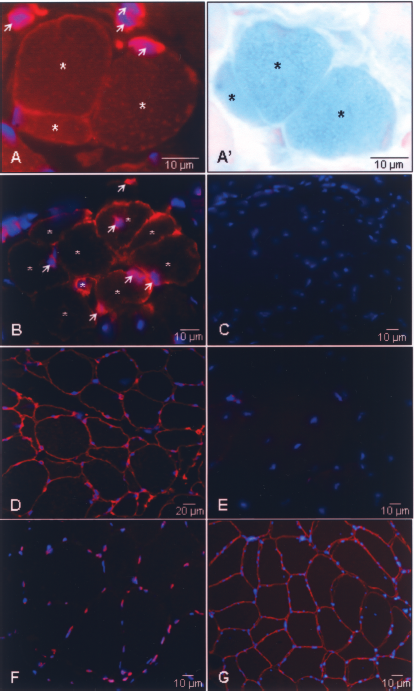
Activation of human skeletal muscle gene expression in HMs. The photographs represent transversal cross sections of CTX-treated TAM collected at 30 days (A) and 60 days (B) after implantation of LacZ-hMSCs. Sections were stained with antibodies specific for human spectrin (sarcolemma, red) and human lamin A/C (nuclear membrane, red) and with the karyophilic dye Hoechst 33342 (all nuclei, blue). The section in (A′) was stained with X-gal (blue) and nuclear fast red (red). The distance between the sections presented in (A) and (A′) is 12 μm. Many human cells (arrowheads) were observed in the vicinity of HMs (asterisks) at 30 (A) and 60 (B) days posttreatment. Notice the clustering of HMs in (B). The control sections consisted of CTX-treated TAM excised 60 days after injury (C), human muscle (D, F, G), and untreated mouse TAM (E). The samples shown in (C), (D), and (E) were incubated with a mixture of the human spectrin- and human lamin A/C-specific antibodies while the sections display in (F) and (G) were only stained for lamin A/C or spectrin, respectively. Magnification (A, A′, B): 1000×; (C, E, F): 630×; (D, F) 400×.
Gradual Loss of hMSCs From Regenerating Murine TAMs
While highly useful to determine the number of HMs, histological analysis of X-gal-stained TAMs treated with LacZ-hMSCs did not allow easy quantification of the mononucleated β-gal+ cells as these often appeared in aggregates. To quantify the total number of human cells in the murine TAMs at different time points after hMSC transplantation we employed the Beta Glo assay for measuring β-gal activity.
Surprisingly, the β-gal activity of CTX-treated TAMs excised 5 min after cell injection (hereinafter referred to as time 0) was much lower than 3 days later and than that of uninjured TAMs (compare Fig. 7A upper and lower graphs). The low values observed early after CTX administration may be due to interference of the strong inflammatory response induced by this myolytic agent with the Beta Glo assay. The first interfering factors that come to mind are proteases released by the infiltrating leukocytes. However, homogenization in the presence of a broad spectrum protease inhibitor cocktail of time 0 CTX-treated TAMs did not result in higher β-gal activity (Fig. 7A, lower graph). Failing to obtain a reliable initial value, we have related the decrease in chemiluminescence over time to the β-gal signal recorded at day 3.

Time-dependent decrease of human cells in TAMs after injection of LacZ-hMSCs. (A) β-gal activity in TAMs. Upper graph: mice treated with CTX only (white bars) and mice receiving LacZ-hMSCs 24 h after CTX injection (black bars) were sacrificed at different time points. β-gal activity per TAM was quantified by the Beta Glo assay and expressed as relative light units (RLUs). Lower graph: β-gal activity in TAMs excised 5 min after implantation of LacZ-hMSCs into CTX-treated and control muscles. Where indicated, a cocktail of protease inhibitors (PIs) was present during the homogenization procedure. (B) β-gal activity in pairs of hind limb muscles following CTX injury of the TAMs and injection of LacZ-hMSCs in one of them. The muscles were collected at 3 or 30 days after cell administration as indicated. The black and white bars correspond to muscles in the hind limbs that did and did not receive donor cells, respectively. EDL, extensor digitorum longus; Sol, soleus; Gas, gastrocnemius.
Comparison of the β-gal activity at day 3 with that at 30 and 60 days after LacZ-hMSC injection revealed a decrease of 79.4% and 89.8%, respectively. This decline was consistent with the histochemical analysis showing a time-dependent decrease of the β-gal+ mononucleated cell population.
Several processes besides donor cell death may account for the reduction of β-gal activity including: (i) epigenetic silencing of transgene expression in the human cell population and (ii) migration of the implanted cells to neighbouring muscles. Although epigenetic silencing of the LacZ transgene cannot be fully excluded, we consider this possibility unlikely because of the use of a CpG islandless reporter gene cassette and the observation that even after more than 3 months of ex vivo culture essentially all LacZ-hMSCs remained strongly β-gal+. As to the migration issue, quantification of β-gal activity in muscles adjacent to the treated TAM at 30 days after injection of LacZ-hMSCs showed low levels in all three tested muscles (Fig. 7B). The total additive β-gal activity did not exceed 5% of the β-gal signal measured in the TAM of the same mice.
Discussion
In this study, we evaluated the capacity of culture-expanded hMSCs derived from BM of an adult donor to participate in murine skeletal muscle regeneration. A CTX-induced myonecrosis model was used to address basic issues, in particular the quantitative and temporal aspects of the regeneration process. These included the intramuscular distribution of locally injected donor cells, their persistence, and their ability to contribute to the formation/repair of murine myofibers at different stages of the healing process.
The CTX from Naja mossambica mossambica used in this study was reported to offer the advantage of selective toxicity. While causing degeneration of myofibers, it does not affect satellite cells, blood vessels, and the myelinated part of motoneurons allowing the regeneration process to start shortly after damage induction (7). We observed that the injection of CTX affected essentially the entire TAM causing necrosis of 90% of the myofibers at 24 h after administration.
The first signs of myoregeneration were observed at 10 days after CTX injection. Remarkable was the ongoing skeletal muscle remodeling at 4 months after injury, as revealed by the presence at this time point of small clusters of myofibers of heterogeneous diameter positioned within the otherwise well-organized fascicles (5,7,53). At the same time point, centrally located nuclei, which are characteristic of regenerating and regenerated skeletal muscle tissue of rodents (33), were seen in the vast majority of myofibers reflecting the massive damage induced by the CTX treatment.
Our results demonstrate convincingly that BM-hMSCs, similar to AT-hMSCs and SM-hMSCs, contribute to myofiber formation/regeneration in mice and persist in murine skeletal muscle tissue for long periods of time (12,50). So far, the only data available for naive BM-hMSCs were limited to 1 week after cell implantation (54). Because regeneration is just beginning at that time after CTX treatment, the extremely low percentage (i.e., 0.02%) of HMs recorded by Shi et al. (54) does not reflect the full potential of these cells. In our study, quantitative histochemical analysis at 1 month after BM-hMSCs injection yielded an average number of 26 β-gal+ myofibers (Fig. 3B) equivalent to ±1% of murine myofibers with human cell contribution. The study on SM-hMSCs (12) does not provide any values for comparison with our data of BM-hMSCs. It merely states that no β-gal+ myofibers were observed at 1 week and “some” at 3 weeks after cell transplantation in CTX-treated TAMs of nude mice. In this context it is of interest that De Bari et al. (12) observed a much higher contribution of SM-hMSCs to the regeneration of TAMs in immunosuppressed mdx mice. At 4 weeks after local injection of 106 SM-hMSCs, they recorded a maximum number of 69 human dystrophin-positive myofibers per transversal TAM section, which is about sevenfold the maximal number of HMs that we scored in one section at the same time point posttransplantation (Fig. 3B, white bars). With regard to hMSCs derived from AT, only few studies are available. Rodriguez et al. (50) performed in vivo experiments in mdx mice with cells that had undergone an unusually large number of population doublings (i.e., 160) in culture. They described up to 50% of the myofibers to be dystrophin positive at 10 days after cell injection and up to 90% at 6 months. If these observations are confirmed, it would indicate that both AT-hMSCs and SM-hMSCs have a much higher myoregenerative capacity than BM-hMSCs (5% HMs at 4 months after injection of 5 × 105 cells) or that dystrophic TAMs provide a more favourable environment for hMSC-dependent skeletal muscle repair than artificially damaged TAMs of nondystrophic mice. A recent study by Goudenege and colleagues (26), who used cells from the same AT-hMSC preparations as Rodriguez et al. (50) but in immunodeficient Rag2–/–γC–/– mice instead of mdx mice, suggests that the latter may indeed be the case. These authors showed that cryodamaged muscles on average contained less than 10 HMs per TAM section at 30 days after injection of naive human AT-MSCs.
Liu et al. (41) also employed mdx mice but in combination with CTX to study the myoregenerative ability of AT-hMSCs. The only information they provide is the observation of “clusters of dystrophin-positive myofibers” at 4 weeks and of “considerable numbers” of dystrophin- and human β-2-microglobulin-positive myofibers at 12 weeks after cell transplantation.
Although our quantitative analysis revealed a fourfold increase in the frequency of HMs per whole TAM between day 30 and 120 after CTX-induced injury, the total number of human cells decreased gradually to reach ±10% of the implanted cells at 60 days after damage induction. This finding, together with the microscopically observed time-dependent reduction in the occurrence of β-gal+ mononucleated cells, may suggest that the CTX-damaged TAM provides a suboptimal microenvironment for long-term maintenance of mononucleated donor cells. In line with this hypothesis, Eliopoulos et al. (17) showed that the survival of murine MSCs following subcutaneous injection into syngeneic recipients was limited but could be greatly increased by embedding the donor cells into a matrix prior to implantation (17,18). In our experimental setting, incorporation into myofibers may also contribute to the maintenance of donor cells.
The finding of β-gal+ mononucleated cells and HMs being distributed over the whole length of TAMs (Fig. 4) implies migration of BM-hMSCs from the site of injection in the center of the TAM to other parts of the muscle. In this context it is interesting that Rodriguez et al. (50) have reported the presence of dystrophin-positive myofibers in the gastrocnemius muscle after injection of AT-hMSCs into the neighboring TAM. Furthermore, our histological screening revealed the presence of isolated HMs dispersed throughout the muscle early after treatment, whereas clusters of β-gal+ HMs were seen at later time points. This may have resulted from late fusion of residual donor cells with regenerating myofibers. Alternatively, it may represent a contribution of donor cell-derived satellite cells as postulated for AT-hMSCs (41) and SM-hMSCs (12).
As previously found in studies focusing on the contribution of muscle precursor cells to myoregeneration (4,42), we identified two types of β-gal-expressing myofibers: short myofibers that stained entirely blue and mosaic myofibers containing blue segments. Our data do not allow concluding whether the fully blue myofibers derive from donor cells only or from their fusion with murine cells. Due to its cytoplasmic localization, diffusion of β-gal throughout myofibers and beyond nuclear domain boundaries (35) cannot be excluded. Other types of analysis that enable the dissociation between human and murine components at the nuclear or cytoplasmic level will be required. Characteristic for the mosaic myofibers was the presence of β-gal+ segments of diverse lengths often staining most intensely in the middle and reaching lengths of up to 2,000 μm (Fig. 5). Similar bell-shaped distributions of β-gal activity have been documented for regenerating myofibers of mdx mice transplanted with cultured myoblasts. The phenomenon was proposed to be due to the fusion of single donor cells to host myofibers (35). The different lengths of the cytoplasmic β-gal+ segments were suggested to be a reflection of an increase in β-gal domain size with time after fusion.
Finally, the detection of human spectrin in HMs at 30 days after cell injection indicated myogenic reprogramming of donor cell nuclei. In this respect, our results with hBM-MSCs are similar to those obtained for hMSCs derived from AT (50) and SM (12).
In summary, we have shown the long-term participation of BM-hMSCs derived from an adult donor in skeletal muscle repair in the mouse. In the CTX-injured muscle model with ongoing regeneration/remodeling, BM-hMSCs and/or their derivatives continue to contribute to skeletal muscle repair resulting in ±5% of HMs at 4 months after transplantation. The expression of endogenous human skeletal muscle genes as well as added transgene by BM-hMSCs after their incorporation into myofibers provided a rationale for their further development into therapeutic tools for DMD.
Footnotes
Acknowledgments
Bone marrow samples for the isolation of MSCs were kindly provided by Simone Hogendoorn and Rob G H. H. Nelissen, Department of Orthopedics, Leiden University Medical Center. We thank Marloes van de Watering (Virus and Stem Cell Biology Laboratory, Department of Molecular Cell Biology, Leiden University Medical Center, the Netherlands) for generating the lentivirus vector. This study was supported by a Scholarship to A. S. de la Garza-Rodea from the Universidad Autónoma de Nuevo León, Monterrey, México and by research grants from the Association Française contre les Myopathies (AFM; 12259-SR-GROUPE E), the Duchenne Parent Project, and the Leiden University Fund (LUF pilot study 7207/5-3-06Kk).