
Abstract
Ensheathing glia have been demonstrated to have neuroregenerative properties but this cell type from human sources has not been extensively studied because tissue samples are not easily obtained, primary cultures are slow growing, and human cell lines are not available. We previously isolated immortalized ensheathing glia by gene transfer of BMI1 and telomerase catalytic subunit into primary cultures derived from olfactory bulbs of an elderly human cadaver donor. These cells escape the replicative senescence characteristic of primary human cells while conserving antigenic and neuroregenerative properties of ensheathing glia, but their low proliferative rate in culture complicates their utility as cell models and their application for preclinical cell therapy experiments. In this study we describe the use of a conditional SV40 T antigen (TAg) transgene to generate human ensheathing glia cell lines, which are easy to maintain due to their robust growth in culture. Although these fast growing clones exhibited polyploid karyotypes frequently observed in cells immortalized by TAg, they did not acquire a transformed phenotype, all of them maintaining neuroregenerative capacity and antigenic markers typical of ensheathing glia. These markers were also retained even after elimination of the TAg transgene using Cre/LoxP technology, although the cells died shortly after, confirming that their survival depended on the presence of the immortalizing genes. We have also demonstrated here the feasibility of using these human cell lines in animal models by genetically marking the cells with GFP and implanting them into the injured spinal cord of immunosuppressed rats. Our conditionally immortalized human ensheathing glia cell lines will thus serve as useful tools for advancing cell therapy approaches and understanding neuroregenerative mechanisms of this unique cell type.
Keywords
Introduction
Olfactory ensheathing glia (OEG) have been intensively studied in recent years due to their neuroregenerative properties [(6,17,38,40,48,49,51,61), recently reviewed by Chiu et al. (9)]. Several groups have demonstrated axonal regeneration in murine spinal cord injury models after ensheathing glia transplantation [(27,29,50), reviewed in Franssen et al. (19)] and some studies have even documented functional recovery (24,28,35,39,45, 46,59) even in humans (5). For future use of this approach in the clinic it is necessary to characterize OEG of human origin: these cells can be obtained either from the olfactory bulb or from the olfactory mucosa. Surgical intervention of the olfactory bulbs is highly invasive and thus culture of bulb-derived glia can only be considered from cadaver donors. In contrast, mucosa-derived OEG of volunteers can be cultured from nasal biopsies, which can be obtained by outpatient surgery (4,12,26, 37,54).
Like many other differentiated human cells, OEG have a limited life span in culture (22,31) which depends on the age of the donor and the culture conditions (20); eventually the cells enter into a nonreplicative state called senescence (25). Many immortalizing strategies have been described in human cell lines [see Lim et al. (30) for a recent review]. Various studies have reported that gene transfer of BMI1 and the catalytic subunit of telomerase (TERT) enables the bypass of senescence of human primary cultures without altering their original properties (11,42). Although we have also found that BMI1/TERT transduction can immortalize human OEG (31), the proliferation rate of these cells is very low, limiting their utility for intensive characterization in vitro or the development of in vivo assays.
Primary culture senescence has been related to the activation of at least two main pathways: those of p16Ink4a/Rb and p19Arf/p53 (8). Extensive literature reports the use of SV40 T antigen (TAg) to immortalize primary cells based upon the ability of this viral protein to bind and inhibit downstream effectors, pRb and p53 family proteins, of both senescence cascades (1).
Reversible immortalization is a technique by which cellular proliferation can be extended by the introduction of transgenes [for a recent review, see (30)], and which, after the cells have been cultured for the desired time, can be silenced or eliminated using temperature-sensitive mutants, ligand-regulated proteins, conditional promoters, or Cre-lox technology. In the latter case, excision of the immortalizing gene is achieved by delivery of Cre recombinase, which catalyzes specific recombination between loxP sites flanking the transgene. Recently, other such cell-reprogramming approaches have been developed to retrodifferentiate primary tissues into induced pluripotent stem (iPS) cells [58), reviewed in (44)], or even transdifferentiate cells of one tissue type directly into another (16,60,62,63). These strategies offer the possibility to generate any cell type from easily obtained tissues such as skin fibroblasts, but considerable effort in these fields is still needed to achieve directed differentiation of iPS cells into specific cell types. Reversible immortalization is an approach in which cell life span can be increased without altering the differentiation stage, thus bypassing the need for directed differentiation.
We previously used Hlox lentivectors for reversible gene transfer of BMI1, TERT, and TAg in a primary culture of ensheathing glia from a 58-year-old male cadaver donor (31). The advanced age of the donor limited the life span in vitro to a few passages and only the BMI1/TERT combination was able to immortalize these cells. However their proliferation rate was very slow, making routine use of these cells difficult. In the present study we show that although TAg was originally unable to immortalize the primary cells of the elderly donor, delivery of TAg into the BMI1/TERT-transduced slow growing cells strikingly improves their growth characteristics without altering OEG properties such as cell type-specific antigenic markers and the capacity to induce axonal regeneration of adult rat retinal ganglion neurons. Additionally, because we also used Hlox lentivectors for reversible TAg gene transfer, this transgene could be efficiently eliminated 1 week after Cre recombinase delivery, leaving marker expression and neuroregenerative capacity intact. In this study we describe several fast growing human ensheathing glia cell lines that offer the potential as useful tools for studying the neuroregenerative properties of this unique glial cell type as well as for testing preclinical cell therapies in vivo in animal models.
Materials and Methods
Special Reagents
MTT (3-[4,5-dimethylthiazo-2-yl]-2,5-diphenyltetrazolium bromide), forskolin, DL-2-amino-5-phosphonovaleric acid, poly-L-lysine (PLL), and polybrene were purchased from Sigma (St. Louis, Mo). Fetal calf serum (FCS) was obtained from Hyclone (Logan, UT). DMEM/F12 medium, pituitary extract, B-27 supplement, Neurobasal medium, and colcemid were purchased from Gibco (Carlsbad, CA). Primocin was from InvivoGen (San Diego, CA). Lipofectamine Plus reagent was from Invitrogen (Carlsbad, CA). Fluoromount G was from SouthernBiotech (Birmingham, AL) and papain was from Worthington's Papain System (Worthington, Lakewood, NJ). To-Pro-3 was purchased from Molecular Probes (Eugene, OR). Dexamethasone (Fortecortín) was from Merck (Barcelona, Spain); cyclosporin A (Sandimmun) from Neoral, Novartis (Barcelona, Spain); ketamine from Pfizer, Park-Davis (Madrid, Spain); and Domtor and Antisedan from Pfizer, Orion Corporation (Espoo, Finland). Tissue-Tek was purchased from Sakura (Zoeterwoude, Netherlands).
Antibodies
Antibodies directed against the listed proteins were used at the indicated dilutions: amyloid precursor protein [immunocytochemistry (ICC) 1/200, MAB348], and nestin (ICC, 1/500, MAB5326), purchased from Chemicon International (Temecula, CA); CRE recombinase [flow cytometry (FC) 1/1000, ab24607], BMI11 (ICC and FC, 1/200, ab14389), and telomerase (ICC and FC, 1/100, ab32020), obtained from Abcam (Cambridge, UK); neuroligin 3 (ICC 1/500, sc-1491) and BMI1 (FC, 1/100, H-99 sc-10745) from Santa Cruz Biotechnology (Santa Cruz, CA); CRE recombinase (ICC and FC, 1/5000, 69050) from Novagen (Gibbstown, NJ); GFP (immunohistochemistry, 1/2000, A11122) from Invitrogen, Molecular Probes (Eugene, OR); microtubule associated protein 2 (MAP2) (ICC, 1/400, 514) as previously described (53); TAg (FC and ICC, 1/250, 554150) from BD Pharmingen (San Diego, CA); phosphorylated neurofilament MAP1B (ICC, 1/500 SMI31) from Sternberger M Inc. (Lutherville, MD); S100β (ICC, 1/500, SH-B1) from Sigma (St. Louis, MO); and vimentin (ICC, 1/200, 814318) from Boehringer (Ingelheim, Germany).
Fluorescent secondary antibodies used for ICC (1/1000) and FC (1/500) were: anti-mouse or anti-rabbit IgGs labeled with Alexa-488, -555, -647, or -594 (Invitrogen).
Lentivector Production and Titration
Pseudotyped lentivectors were produced by cotransfection of 5 μg of the corresponding lentivector plasmid, 5 μg of the packaging plasmid pCMVdR8.74 (14), and 2 μg of the plasmid pMD2G (Addgene plasmid 12259 encoding vesicular stomatitis virus G envelope protein) in 10-cm plates of subconfluent 293T cells using Lipofectamine Plus reagent following instructions of the supplier (Invitrogen). Lentivectors expressing immortalizing genes were pLOX-CWBmi1 (Addgene plasmid 12240), pLOX-TERT-iresTK (Addgene plasmid 12245), and pLOX-Ttag-iresTK (Addgene plasmid 12246), which respectively encode mouse BMI1, TERT coupled to the HSV-1 thymidine kinase (TK) gene via the encephalomyocarditis virus internal ribosome entry site (IRES), and TAg coupled to IRES-TK (52). To express CRE recombinase we used pLOX-CW-CRE (11), a lentivector that transduces cells with a self-excising CRE transgene (LvCRE), thus avoiding genotoxicity due to sustained Cre expression. The pRRLSIN.cPPT.PGK-GFP.WPRE vector (Addgene plasmid 12252), which expresses enhanced green fluorescent protein (E-GFP), was used as a control vector (LvGFP). Lentivectors were titered on target cells (hOEG) with serial dilutions of the vector supernatants and the number of transduced cells determined 48 h postinfection by flow cytometry (FACSCalibur, BD Biosciences, San Diego, CA) using antibodies directed against the corresponding transgene protein.
Culture, Immortalization, and Deimmortalization of Human OEG
The human OEG used in the present study are all derived from our previous study (31) in which the isolation and characterization of the glia have been described in detail. Briefly, we used a previously described method (41) with modifications (31) to obtain OEG primary cultures from adult human olfactory bulbs that were obtained from the Tissue Bank for Neurological Research of Madrid. OEG were identified in the olfactory bulb following established morphological and anatomical guidelines (34) and cultured in ME medium: DMEM/F12 (1:1), 10% FCS, 2 mM glutamine, 20 μg/ml pituitary extract, 2 μM forskolin, 50 μg/ml primocin. Our previous study (31) also describes the infection of these human OEG with lentivectors encoding BMI1 and TERT, isolation of immortalized clones, and the characterization of the clonal lines as well as the primary cells for the expression of glial antigenic markers including S100β, GFAP, neuroligin-3, APP, vimentin, and nestin. One of these BMI1/TERT-expressing OEG clones (hTL4) was infected with a lentivector encoding TAg at a MOI of between 2 and 10, generating the cell lines of the present study, which preserve the antigenic markers of the original cells. Different clones expressing TAg were selected and their proliferation rate was calculated as the slope of the line of best fit of the plot of cumulative population doublings (PD) as a function of time (in days). We calculated PD = log(Nf/N0)/log 2, where Nf is the final cell count and N0 is the initial number of cells seeded. Microsoft Excel was used to perform linear regression and calculate the standard error of the slope. Genetic reversion of immortalized human OEG was carried out by infecting the cells with a lentivector encoding Cre recombinase while a lentivector encoding E-GFP (LvGFP) was used as a negative control. Deimmortalized cells were analyzed for viability using the MTT assay 120 h after infection as previously described (43).
Flow Cytometry Analysis
Cells were detached by trypsin treatment, fixed by 10-min incubation in 2% paraformaldehyde (PFA) in phosphate-buffered saline (PBS), and washed and maintained in PBS at 4°C until analyzed. Immediately prior to analysis, cells were permeabilized by 2-min treatment with PBS + 0.5% Tween 20, followed by blocking with PBS + 0.5% Tween 20 + 10% FCS for 15 min. The cells then were incubated with the primary antibody diluted in blocking solution for 20 min. After washing twice with PBS + 0.5% Tween 20, cells were incubated for 10 min with the secondary antibodies. After washing again, flow cytometry analysis was performed (FACSCalibur, BD Biosciences, San Diego, CA). The negative gate was fixed for each experiment by using the same immunodetection conditions with OEG cells that did not express the protein of interest (TAg or CRE). The positive fraction for each antigen was determined as the percentage of events with higher fluorescence intensity than the negative gate threshold with respect to the total of events plotted.
Cocultures of Postmitotic Rat Retinal Ganglion Neurons with OEG
Extension of neurites by postmitotic adult rat retinal ganglion neurons was used as a model of regeneration in culture (41). Briefly, retinal tissue was extracted from 2-month-old (P60) rats and digested with papain (20 U/ml) in the presence of 50 μM of the NMDA receptor inhibitor DL-2-amino-5-phosphonovaleric acid. The cell suspension was then plated either on 10 μg/ml poly-L-lysine (PLL)-treated coverslips or OEG monolayers. The cultures were maintained at 37°C with 5% CO2 in serum-free Neurobasal medium supplemented with B-27 12.5 mM KCl for 96 h before fixing with 4% PFA.
Quantification of Axon Regeneration
Preparations were quantified in a blinded manner by counting axons under a 40× objective of an inverted Axiovert 200 microscope. A minimum of 20 randomly chosen fields were quantified for each treatment and experiments were performed once (Th1, Tm10, and Ts11) or six times (poly-L-lysine, hTL4, and Ts14). Axonal regeneration was quantified as the percentage of neurons with an axon, whereas total neuron number was determined by staining with the microtubule-associated protein (MAP) 2 antibody. Axons were defined as polarized neurites stained with antibodies against phosphorylated MAP1B and the high molecular weight neurofilament subunit proteins. We also determined the axonal regeneration index, a parameter defined as the axonal length/field as well as the percentage of neurons that regenerate their axons. Quantitative image analysis was performed using the NeuronJ plugin (E. Meijering) of the ImageJ program (Wayne Rasband, National Institutes of Health, USA).
Karyotype Analysis
Chromosomal studies were performed at the Genetics Department of the Hospital “La Paz” in Madrid. Metaphase spreads were prepared from cells treated with 100 ng/ml colcemid for 6 h and analyzed by standard protocols for high-resolution GTL banding (55).
Anchorage Independent Growth
Cells were seeded in 12-multiwell plates at 104 cells/well in 0.35% agar in DMEM culture medium over a 0.7% agar layer. Plates were incubated for 23 days, after which time colonies were large enough to be visualized by staining with MTT (200 μg/ml) for 6 h and photographed using a flatbed scanner. Soft agar assays were performed in triplicate.
Immunocytochemistry
Cells were grown on sterile glass coverslips and fixed with 4% PFA in PBS. After blocking with PBS containing 1% FCS, 0.1% Triton X-100 for 30 min, and an extra permeabilization of 10 min in PBS containing 0.1% SDS for nuclear antigens, cells were washed with PBS and stained by indirect immunofluorescence using the antibodies described above. Samples were mounted in Fluoromount G and were observed in an Axiovert 200 (Zeiss, Oberkochen, Germany) fluorescence microscope or in a Radiance 2000 confocal system (Bio-Rad, Hercules, CA) coupled to an Axiovert S100 TV inverted microscope (Zeiss).
Colony Formation Assay
Ts14 cells were infected at different MOI (0, 1, 2.5, and 10) with lentivectors encoding Cre recombinase (LvCRE) or E-GFP (LvGFP) as control. After 48 h, cells were trypsinized, counted, and 1,000 cells were seeded in six-multiwell plates. After 15 days, plates were stained with methylene blue 1% and the number of colonies was counted.
Animal Treatment
All animal experimentation was carried out in animal facilities (Institutional Registration No. 28079-19A, Spanish Ministry of Agriculture) complying with the Spanish Royal Decree 223/1988, which follows the European Council Directive 86/609/EEC (1986), and approved by national and institutional bioethics committees.
To study tumorigenicity of Ts14 cells, six 12-week-old Hsd: Athymic Nude-Foxn1nu mice (Harlan Interfauna Iberica, Barcelona, Spain) were subcutaneously inoculated with 106 cells in the flanks. As positive controls, six similar inoculations were performed using U-87 MG glioblastoma cell line [ATTC number HTB-14, (3)] while two animals were left untreated as negative controls. The development of tumors was followed up for at least 6 weeks.
To study the survival of Ts14 cell xenografts, male 2-month-old Wistar rats weighing approximately 250 g were treated with dexamethasone (from 2 weeks before cell therapy until euthanasia, 2 mg/L in drinking water, estimated as 30 ml/day) or cyclosporine A (15 mg/kg/day intraperitoneally from 3 days before cell therapy) or dexamethasone (in the same conditions) combined with cyclosporine A (15 mg/kg/day in drinking water from 3 days before cell therapy), n = 3 per treatment group. Rats were operated on under anesthesia (intraperitoneal injection 80 mg/kg ketamine and 0.32 ml/kg Domtor) to produce a bilateral lesion in the dorsal columns at spinal level C3 using watchmaker's forceps (7). To transplant Ts14-GFP cells, a total of 1.5 × 105 cells (1 × 104 cells/μl) were injected with a Hamilton syringe coupled to a glass micropipette, into the dorsal half of the spinal cord (depth 2 mm) at three rostrocaudal levels: at the lesion site and 1 mm caudal to and rostral to the lesion (5 μl/min/site; a total of 15 μl). Rats were reanimated by intraperitoneal injection of 80 ml/kg of Antisedan. After the indicated times rats were sacrificed and tissues were fixed by transcardial perfusion. The spinal cords were dissected out of these animals and embedded in Tissue-Tek and quick frozen in dry ice. Serial cryostat sagittal sections (20 μm) were prepared, immunohistochemistry for E-GFP was performed (using the above described immunocytochemistry protocol), and sections were observed under an Axiovert 200 (Zeiss, Oberkochen, Germany) fluorescence microscope.
Statistical Analysis
Analysis of variance (ANOVA) was performed to test the differences between experimental factors and their interaction. If the differences were significant, Bonferroni's post hoc test for multiple comparisons between means was carried out.
Results
Increased Proliferation of hTL4 by TAg Addition
In our previous study (31) using primary cultures derived from human olfactory bulbs of a 58-year-old donor we were unable to obtain immortalized OEG using lentivectors expressing TAg either alone or in combination with TERT, but, curiously, we did obtain OEG immortalized by BMI1 and TERT. These cells could be cloned and expanded for at least 20 duplications, permitting our characterization of their antigenic profile as well as their neuroregenerative properties in culture assays (31), but their slow growth rate (duplication time of approximately 10 days), compromises their ease of use as cell models or for developing therapeutic approaches in animal models.
We thus explored the ability of a lentivector encoding SV40 large T antigen (TAg) to increase the proliferative rate of these BMI1/TERT-immortalized OEG. We initially analyzed four clones selected according to their different replication rates (Table 1): Th1 with a high replication rate, Tm10 with an intermediate replication rate, and Ts11 and Ts14 with slower replication rates. The mean TAg expression level of each clone, as quantified by flow cytometry (Fig. 1A), did not appear to correlate with proliferative rate (compare with Table 1). Transduction by TAg provoked a significant morphological change: whereas human OEG transduced by BMI1/TERT are large and flattened cells (Fig. 1B), TAg expressing clones are smaller, fibroblast-like, and divide until reaching confluence (Fig. 1C).

Effect of SV40 T antigen on human olfactory ensheathing glia cell lines. Human OEG immortalized originally by BMI1 and TERT (BMI1/TERT) were subsequently infected with a lentivector encoding SV40 T antigen. (A) Analysis of T antigen expression by flow cytometry of four different clones of BMI1/TERT-immortalized cells transduced by TAg (Th1, Tm10, Ts11, and Ts14). Graphs show means and SDs of fluorescence intensity. (B) Representative phase contrast image of BMI/TERT-immortalized hOEG and (C) one TAg-expressing clone Ts14. Scale bars: 100 μm. (D, E) Neuroregenerative capacity of TAg-accelerated OEG cell lines compared with the BMI1/TERT-immortalized parental clone in coculture assays with adult rat retinal ganglion neurons. Neuroregeneration was scored by determining the percentage of neurons extending axons (D) as well as by measuring a parameter proportional to the average length of regenerated axons (E). The graphs show means and SDs of six independent experiments with poly-L-lysine (PLL), BMI TERT, and Ts14; the rest of the clones were measured once. ***p < 0.001 (ANOVA and post hoc Bonferroni test, with respect to PLL).
Proliferation Rate of Each Population

Values represent the slope of the line of best fit of the plot of cumulative population doubling (PD) as a function of the time in days, PDs = log (Nf/N0)/log 2, where Nf is the final cell count and N0 is the initial number of cells seeded. The standard error of the proliferation rate (slope of the line of best fit) is shown.
We next evaluated if TAg expression alters OEG regenerative properties, by comparing the capacity of the four TAg-transduced cell lines to induce axonal regeneration in adult rat retinal ganglion neurons (41), with that of the parental BMI1/TERT-immortalized line (hTL4). The percentage of neurons with an axon (Fig. 1D) was similar in one of the clones (Ts14) and slightly decreased in the other three (Th1, Tm10, Ts11). Indeed, in the case of Ts14 the axonal regeneration index was slightly higher and we thus chose this clone for more detailed studies (Fig. 1E).
Karyotype Stability and Transforming Potential
To study genomic stability after OEG transduction, a high resolution GTL banding assay was performed (Fig. 2A, Table 2). Untransduced OEG showed a normal male diploid karyotype without any alteration in all metaphases analyzed. Some chromosomal instability was evident after BMI/TERT immortalization: all hTL4 cells lacked the Y chromosome and two of the metaphases analyzed showed the additional absence of chromosome 21 or 22. TAg transformation markedly increased chromosome instability: all Ts14 cells were polyploid with varying numbers of chromosomes and multiple rearrangements. Interestingly, these gross karyotypic alterations seem to have minimal effect on the neuroregenerative capacity of Ts14 cells compared to that of the parental line hTL4 as well as that of the original primary OEG cells (31).

Karyotype analysis and anchorage-independent growth of human olfactory ensheathing glia cells. (A) Representative high-resolution GTL-banded metaphase spreads prepared from primary hOEG (control) and those immortalized by BMI1/TERT or BM1/TERT/TAg (Ts14 clone). Quantitative data are given in Table 2. Arrows indicate gross deletions and A, aberrant chromosomes. (B) Representative images of anchorage-independent growth in soft agar of these cells using the human glioblastoma cell line U-87 MG as a positive control.
Karyotype Analysis

To test the transforming potential of these immortalized cells we studied their capacity for anchorage-independent growth. None of these cells was able to form colonies in soft agar (Fig. 2B) in the same conditions as our human glioblastoma-positive control cell line U-87 MG. It thus appears that in spite of substantial karyotypic abnormalities, Ts14 cells do not exhibit tumorigenic behavior. Additionally, six immunodeficient nude mice were inoculated with Ts14 cells in the flanks and none of them have developed tumors at least during the first 6 weeks of an ongoing experiment. In contrast, six out of six inoculations using the human glioblastoma cell line U-87 MG developed noticeable tumors in the first week.
Deimmortalization of Ts14
Because all of the immortalizing genes transduced in our OEG cell lines were flanked by loxP sites (52), they could be excised by Cre recombinase delivery to the cells. To achieve this “deimmortalization,” we used a self-excising Cre lentivector (11), thus avoiding long-term Cre recombinase expression, which may have detrimental genotoxic effects (36,56). As shown in Figure 3A, TAg expression could be eliminated from a population of TAg-transduced hTL4 cells by infection with a Cre-encoding lentivector, whereas infection with a control (GFP-expressing) lentivector under the same conditions had no effect on TAg expression. Immunocytochemical analysis of Ts14 cells confirmed the nuclear localization of BMI1, TERT and TAg, and elimination of their expression with the appearance of Cre in the nucleus (Fig. 3B). Flow cytometry analysis showed TAg expression was eliminated from Ts14 cells positive for Cre, but not GFP (Fig. 3C) and that the percentage of cells that lost TAg expression was proportional to the dosage of LvCre lentivector but unaffected by LvGFP lentivector (Fig. 3D). Some of the cells that are Cre negative also show loss of TAg, due to the self-excising property of the Cre-encoding vector (Fig. 3C).

Cre recombinase-mediated excision of TAg from immortalized human olfactory ensheathing glia. (A) Kinetics of elimination of TAg expression. TAg-transduced OEG originally immortalized by BMI1/TERT were not infected (uninf) or infected with a lentivector encoding CRE (LvCRE) or a control lentivector carrying E-GFP (LvGFP) at a MOI of 10 and at the indicated times. TAg expression was monitored by flow cytometry. (B) Representative immunofluorescence images showing BMI1, TERT, TAg, and CRE expression in Ts14 cells before and after deimmortalization with LvCRE (MOI: 1–10). Scale bars: 20 μm. (C) Flow cytometry dot plots representing TAg expression on the x-axis and Cre recombinase or GFP expression on the y-axis. Ts14 cells not treated with vectors (control) or infected with lentivectors enconding Cre recombinase (LvCRE, right column) or E-GFP (LvGFP, left column) at the indicated MOI. (D) Graphs showing quantification of the experiment detailed in (C). Cells positive for TAg expression: right gates; cells positive for CRE or GFP: upper gates.
Having confirmed that Cre expression in Ts14 cells eliminates expression of the transgenes, thus genetically reverting these immortalized human OEG cells, we next characterized the biological effect of this “deimmortalization.” Strikingly, while treatment with LvGFP vector did not significantly affect the cells, viability (assayed 5 days after infection) was drastically reduced by treatment with LvCre vector (Fig. 4A) in a dose-dependent manner (Fig. 4B). The effect of deimmortalization on the long-term survival of Ts14 cells was also studied by plating the cells at high dilution and observing colony formation after 15 days (Fig. 4C, D). Again, the survival of Ts14 cells was reduced in a dose-dependent manner by the LvCre vector but not by the LvGFP vector, although a slight toxic effect (probably due to the lentivector itself) was observed in this assay (Fig. 4D; compare LvGFP open bars with control gray bar). Our results thus indicate that Ts14 proliferation depends on the presence of the immortalizing transgenes and the cessation of cell division when these are removed provokes cell death.
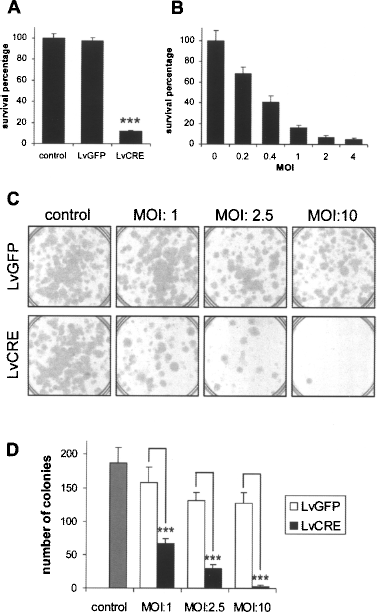
Cell death of Ts14 cells after deimmortalization by Cre-mediated transgene excision. (A, B) Viability of Ts14 cells was measured by the MTT assay 120 h after infection with lentivectors. Cell viability is expressed as a percentage of uninfected cell (control) values and the graphs display the means and SDs of three independent samples. ***p < 0.001 (ANOVA and post hoc Bonferroni test). (A) Cell viability was severely compromised by infection with LvCRE vector while an equal amount of LvGFP vector had no significant effect. (B) Infection with LvCRE at the indicated various MOI showed a dose-dependent effect on cell viability. (C) Representative images of the colony formation assay of Ts14 cells infected with LvGFP or LvCRE lentivectors at the indicated various MOI. (D) Quantification of colony number in the assay shown in (C). The graph depicts the means and SDs of three different samples. ***p < 0.001 (ANOVA and post hoc Bonferroni test).
Using immunofluorescence we also characterized Ts14 cells for the expression of typical OEG markers (Fig. 5) as well as the effect of deimmortalization on these markers. Similarly to primary human OEG, Ts14 cells stained positively for S100β, APP, neuroligin 3, nestin, and vimentin both before and after infection by the LvCre vector, showing that neither TAg transduction nor transgene elimination modified the OEG antigenic profile of these cells compared with the parental cell line (31).

Immunocytochemical analysis of Ts14 cells. Representative images of immunofluorescence staining for characteristic OEG markers in Ts14 cells before and after deimmortalization by LvCRE infection (MOI: 10). Similar to primary human OEG (31), Ts14 cells exhibited immunostaining for the markers S100β, APP, neuroligin-3, nestin, and vimentin, and expression of these markers was not altered by deimmortalization. Scale bar: 20 μm.
Xenografts of Ts14 for In Vivo Evaluation of Human OEG Cell Therapy
The development of human cell-based therapies is fraught with practical problems such as the lack of large amounts of homogenous, precharacterized graft tissue, and viable in vivo models in which to perform preclinical trials. Our human OEG cell lines offer one way to obtain enough graft material of known characteristics, so we next investigated the possibility of using these cells as xenografts in a rat model of spinal cord lesion (39). To trace these cells in vivo we labeled the Ts14 cell line by infecting them with a lentivector expressing E-GFP (LvGFP, MOI: 10) and selecting a clone (Ts14G) expressing easily detectable levels of GFP by flow cytometry. These cells were transplanted into rat spinal cords subjected to dorsal column crush at C3 level. Although transplanted Ts14G cells could be clearly visualized 24 h after transplantation (Fig. 6A), none could be detected 1 week after inoculation, probably due to a xenograft-induced immune response (Fig. 6B). To test this hypothesis we studied the effect of immunosuppressive treatments: animals were treated with dexamethasone from 2 weeks before transplantation and/or cyclosporine A until the day of sacrifice. Using any of these three immunosuppressive protocols, Ts14G cells could be clearly visualized 1 week after transplantation (Fig. 6C–E) and in some cases positive cells could even be detected 4 weeks after transplantation (Fig. 6F). These results encouragingly indicate that these human OEG cell lines can be used for in vivo studies in rodents, even when transplanted into an immunologically hostile environment such as the crush-lesioned spinal cord. Further experiments using immunodeficient animals may extend the survival of these cells to enable important long-term functional studies of human OEG-mediated neuroregeneration in vivo.

Effect of immunosuppression on persistence of human Ts14 cells transplanted into a rat model of spinal cord lesion. After dorsal column crush at the level of C3 in Wistar rats, Ts14G cells (1.5 × 105 cells distributed equally in three injections) were innoculated into the lesion site. Three animals were used for each treatment. Representative images shown are of GFP immunofluorescence staining of spinal cord saggital sections. Untreated rats 24 h (A) and 1 week (B) after transplantation. Immunosuppressed rats 1 week after transplantation treated with dexamethasone (C), cyclosporine A (D), or both (E), or 4 weeks after transplantation treated with cyclosporine A (F). Scale bars: 200 μm (A) or 500 μm (B–F).
Discussion
The generation of neural cell lines from human primary cultures greatly facilitates the characterization of these cells as well as their development for use for cell-based therapies. Depending on cell type, culture conditions and donor age primary cultures have varying replicative life spans, after which they enter a nonreplicative state called senescence (25). Immortalization offers the possibility to increase the life span of cells, thus enabling the amplification of scarce primary neural tissue. The production of immortalized stocks also assures a homogeneous source of cells which can be precharacterized to generate models to enable the comparative research that is necessary for a cell therapy product.
Although different combinations of immortalizing genes, such as BMI1 and TERT (31), are able to immortalize OEG cells, the proliferation rates are low, making very difficult their characterization and use in animal experiments. In this work we have demonstrated that TAg efficiently increases proliferation rate of these previously immortalized cells while maintaining their lineage specific markers and neuroregenerative properties.
Immortalization can be considered as the bypass of senescence, which is the consequence of cell aging in culture conditions. Two main pathways are involved in this process: those of p16/pRB and p19/p53/p21 (8). Successful bypass of senescence depends on the ability of the immortalizing genes to repress both pathways (13) as well as the preexisting cellular levels of the proteins involved in these pathways (2). Indeed, we previously demonstrated that OEG from an elderly donor could be immortalized by the BMI/TERT combination but not by using TAg either alone or combined with TERT (31), probably because TAg is unable to block senescence progression in this cell type (22). However, our present study indicates that once the senescence process is inhibited by the BMI1/TERT combination, TAg is able to efficiently increase the proliferation rate.
Our present data show that deimmortalization of TAg-accelerated BMI1/TERT cells not only results in cessation of proliferation but also in cell death. This offers a potential tool for eliminating these cells in animal studies to demonstrate the causative role of OEG in neuroregeneration. Furthermore, this mechanism could be employed to improve biosecurity in cell therapy strategies for the removal of unwanted graft cells. Relevant to these applications, several methods for controlling Cre activity have been developed as biological switches: 1) vectors in which transgene expression is controlled by small nontoxic molecules such as tetracycline (23,57), ecdysone (21,47), or tamoxifen (15,33); 2) direct protein delivery of a cell-permeant Cre recombinase (32). Finally, strategies to achieve cell type specific gene transfer (18) could be applied to target Cre delivery to the engrafted cells.
It is important to note that TAg-induced karyotypic instability and this raises the possibility of other uncontrolled events, which could eventually immortalize the cells independently of TAg, thus rendering the cells insensitive to deimmortalization. In our hands, however, we have not observed such resistant mutants, and the loss of TAg appears to consistently reactivate the p53 pathway that induces the S-phase cell cycle check point to detect gross chromosome abnormalities and induce cell death.
We have demonstrated that Ts14 cells can be successfully implanted in immunosuppressed rats and traced for 1 month and, to date, with no signs of tumorigenicity. Although further studies in immunocompromised animals are necessary to prolong graft persistence to enable functional recuperation measurements to be performed, these preliminary results reveal the potential of this model not only to study the neuroregenerative capacity of the ensheathing glia cell in spinal cord injury models but also to study their migration in the lesion as well as their interaction with host cells.
Although there is increasing knowledge about reprogramming of mature somatic tissues to generate induced pluripotent stem cells (iPS) for large-scale expansion in culture, the complex task of the correct differentiation of these cells to the desired tissue remains to be achieved. Relevant to this point, mesenchymal stem cells have been used in spinal cord injury models (10), but it is still unclear how to control the differentiation of such adult stem cells into specific neural cell types. In this sense, the differentiation process can be altogether avoided by using reversibly immortalized cells such as Ts14, which offer the possibility to efficiently obtain expanded primary tissues without altering the desired properties of the cells.
Footnotes
Acknowledgments
We are grateful for advice and technical aid provided by the Microscopy service of the Centro de Biología Molecular “Severo Ochoa.” This work was supported by grants from Noscira S.A. and the Fundación Marcelino Botín. Filip Lim held Ramón y Cajal and I3 research incorporation contracts from the Spanish Ministry of Science.