
Abstract
This in vitro study describes a novel cell culture, transport, and transfer protocol that may be highly suitable for delivering cultured proliferating keratinocytes and melanocytes to large open skin wounds (e.g., burns). We have taken into account previous limitations identified using other keratinocyte transfer techniques, such as regulatory issues, stability of keratinocytes during transport (single cell suspensions undergo terminal differentiation), ease of handling during application, and the degree of epidermal blistering resulting after transplantation (both related to transplanting keratinocyte sheets). Large numbers of proliferating epidermal cells (EC) (keratinocytes and melanocytes) were generated within 10–14 days and seeded onto a three-dimensional matrix composed of elastin and collagen types I, III, and V (Matriderm®), which enabled easy and stable transport of the EC for up to 24 h under ambient conditions. All culture conditions were in accordance with the regulations set by the Dutch Central Committee on Research Involving Human Subjects (CCMO). As an in vitro model system for clinical in vivo transfer, the EC were then transferred from Matriderm onto human acellular dermis during a period of 3 days. After transfer the EC maintained the ability to regenerate into a fully differentiated epidermis containing melanocytes on the human dermis. Proliferating keratinocytes were located in the basal layer and keratin-10 expression was located in differentiating suprabasal layers similar to that found in human epidermis. No blistering was observed (separation of the epidermis from the basement membrane). Keratin-6 expression was strongly upregulated in the regenerating epidermis similar to normal wound healing. In summary, we show that EC-Matriderm contains viable, metabolically active keratinocytes and melanocytes cultured in a manner that permits easy transportation and contains epidermal cells with the potential to form a pigmented reconstructed epidermis. This in vitro study has produced a robust protocol that is ready for clinical studies in the future.
Introduction
Optimal closure of large cutaneous wounds such as burns, trauma-induced wounds, and surgical excision wounds is still a major challenge for clinicians. Delayed wound closure increases the risk of infection, dehydration, and adverse (hypertrophic) scar formation. Therefore, the time taken for wound closure is very important for the final quality of the wound healing. Transplantation of full or split-thickness (often meshed) autograft is still the most commonly used method for closing large cutaneous wounds (2). However, results are often considered suboptimal, particularly when expanded meshed autografts are used. Also, when treating particularly large wounds, the availability of donor site skin may be a limiting factor. Alternative methods for closing large wounds and in particular for reepithelializing these wounds have been sought for many years. It is thought that application of cultured epithelial cells in combination with a dermal matrix may provide a future means of obtaining superior wound healing while at the same time solving the problem when donor site skin is limited.
Application of cultured confluent autologous keratinocyte sheets was the first alternative method used to replace split-thickness autograft, particularly for the treatment of major burns. The method was developed by Rheinwald and Green in 1975 (22) and the first application for treating extensive third-degree burns was in 1981 (20). Whereas at the time this methodology provided a life-saving breakthrough for many patients, a number of major drawbacks have now been realized, making it unacceptable for current medical and ethical testing committees. These drawbacks include the use of lethally irradiated 3T3 murine fibroblasts in order to sufficiently amplify the yield of keratinocytes. Such coculture is accompanied with the risk of viruses, prions, or other macromolecules being transmitted from the feeder cells to the human cells during culture (5). In a previous study we solved this problem by culturing keratinocytes on collagen IV, which eliminated the requirement of feeder layer cells while still maintaining a satisfactory amplification factor (7).
Other drawbacks with regard to the original Rheinwald and Green method include the use of high concentrations of bovine serum in the culture medium, which results in noncompliance with current regulatory requirements; the extreme fragility of the sheets, which makes transport logistics and application onto the wound surface very difficult, and importantly the enzymatic (dispase) digestion required to remove the sheets from the tissue culture plastic, which results in poor and highly variable take, and frequent blistering of the sheets after application (2,3,6,8,13,14). In this study, we have taken all of these previous drawbacks into account and have developed a means to amplify large numbers of proliferating epidermal cells (EC) (keratinocytes and melanocytes) and a means to easily transport and finally transfer the cells onto a dermal matrix, all in accordance with the regulations set by the Dutch Central Committee on Research Involving Human Subjects (CCMO). This in vitro study shows that a three-dimensional (3D) matrix composed of elastin and collagen types I, III, and V (Matriderm®) can be used as an effective transport carrier system to enable easy transport and transfer of amplified, proliferating EC (keratinocytes and melanocytes) onto human acellular dermis. After transfer we show that the EC can then regenerate to form a fully differentiated epidermis containing melanocytes.
Materials and Methods
Epidermal Cell Culture
Human adult skin was obtained from healthy donors (with informed consent) undergoing abdominal dermolipectomy and was used directly after surgery. The VU University Medical Center approved of the experiments described in this article.
From 3 cm2 full-thickness skin, epidermal sheets were separated from dermis by incubation in Dispase II (Roche, Mannheim, Germany) overnight at 4°C. Keratinocytes and melanocytes were isolated from the epidermis by incubating for 10 min in trypsin 0.125% (Hyclone, UT, USA). Cells were then co-seeded into tissue culture dishes coated with 0.5 μg/cm2 human placental collagen IV at a density of 3 × 106 cells per 9-cm-diameter culture dish (Sigma-Aldrich, St. Louis, MO, USA) and cultured in keratinocyte culture medium at 37°C, 7.5% CO2. Keratinocyte culture medium consisted of Dulbecco's modified Eagle's medium (DMEM)/ Ham's F12 (Lonza, Verviers, Belgium) in a 3:1 ratio supplemented with 5% fetalclone III (Hyclone), 1 μmol/L hydrocortisone (Sigma-Aldrich), 1 μmol/L isoproterenol hydrochloride (Sigma-Aldrich), 0.09 μmol/L insulin (Sigma-Aldrich), 2 ng/ml human keratinocyte growth factor (Sigma-Aldrich), 50 U/ml penicillin, and 50 U/ml streptomycin (Lonza, Verviers, Belgium). The culture medium was accepted by the Dutch Central Committee on Research Involving Human Subjects (CCMO) as suitable for culturing epidermal cells for use in human clinical studies.
After approximately 10–14 days of culture in which the cultures were passaged twice, epidermal cells (keratinocytes and melanocyte coculture) were seeded at a density of 50,000 cells/cm2 onto Matriderm, a 10 × 15 cm collagen–elastin matrix (dr. Suwelack Skin & Health Care, Billerbeck, Germany), which has the registration as a medical device. Epidermal cell-loaded Matriderm (EC-Matriderm) was further cultured at 37°C and 7.5% CO2 for 3–5 days. For quality control, 3-mm-diameter biopsies were taken from the EC-Matriderm for analyses by MTT assay (at least three biopsies were taken from each of six independent donor experiments) and for (immuno)histological analysis. From three of these experiments, 1-cm2 biopsies were also taken from each EC-Matriderm in order to determine the transferability and regenerative potential of the epidermal cells (three different donors, in duplicate). A schematic diagram of the protocol is shown in Figure 1.

Schematic diagram of the culture protocol.
Transport Conditions for EC-Matriderm
Three days after seeding EC onto Matriderm, the EC-Matriderm was washed twice in Hank's balanced salt solution (HBSS) (Lonza, Verviers, Belgium). Excess fluid was removed and the EC-Matriderm was left under ambient transport conditions (20°C, atmospheric oxygen and humidity) for 24 h. After 24 h, the EC-Matriderm received fresh keratinocyte culture medium and was transferred to the culture incubator for a further 48 h in order to determine whether the 24-h transport condition had long-lasting detrimental effects on cell metabolism and survival. Biopsies (10 × 3 mm diameter) were taken from different areas of the EC-Matriderm at time 0 (control), after 24 h at ambient condition, and after 48-h replacement in the culture incubator. Biopsies were analyzed by MTT assay to determine metabolic activity and cell survival. The values of the 10 biopsies obtained per experiment were averaged. The experiment was performed with three different donors, each in duplicate.
Transfer of Epidermal Cells From EC-Matriderm Onto Human Dermis and Regeneration of the Epidermis
Acellular human dermis was prepared from glycerolpreserved donor skin obtained from Euro Skin Bank, (Beverwijk, The Netherlands) by repeated washing in phosphate-buffered saline (PBS) as previously described (27). EC-Matriderm (1 cm2) was washed for 1 h with HBSS and then laid onto the acellular dermis. After 3 days, EC-Matriderm was removed and the dermis with the transferred epidermal cells was cultured for a further 2 weeks at the air–liquid interface. Culture medium consisted of DMEM/Ham's F12 in a 3:1 ratio supplemented with 1% fetalclone III, 1 μmol/L hydrocortisone, 1 μmol/L isoproterenol hydrochloride, 0.1 μmol/L insulin, 4 ng/ml keratinocyte growth factor human, 50 U/ml penicillin and 50 U/ml streptomycin, 10 μmol/L l-carnitine hydrochloride (Sigma-Aldrich), 0.01 mol/L l-serine (Sigma-Aldrich), 1 ng/ml epidermal growth factor (Sigma-Aldrich), 1 μmol/L dl-α-tocopherol (Sigma-Aldrich), and 0.4 mmol/L l-ascorbic acid (Sigma-Aldrich), enriched with a lipid supplement containing 7 μmol/L arachidonic acid (Sigma-Aldrich), 25 μmol/L palmitic acid (Sigma-Aldrich), 15 μmol/L linoleic acid (Sigma-Aldrich), and 24 μmol/L bovine serum albumin (Sigma-Aldrich). Hereafter, cultures were harvested for analysis of cell viability (MTT assay) and (immuno)histology.
MTT Assay
The MTT assay measures mitochondrial metabolic activity. This assay is based on the cleavage of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] to formazan crystals by metabolic active cells. The amount of formazan crystals directly correlates to the number of viable cells. Formazan crystals are solubilized and the resulting colored solution is quantified using an ELISA reader. In short, 3-mm biopsies of EC-Matriderm or reconstructed epidermis on dermis were transferred to a 96-well flat-bottom microtiter plate filled with 200 μl per well of 2 mg/ml MTT labeling reagent (Roche, Penzberg, Germany). After incubation for 2 h at 37°C the biopsies were transferred to a 96-well microtiter plate containing 200 μl per well isopropanol, acidified with 0.04 M HCL (3:1) and incubated overnight at room temperature. Extractant solution (100 μl) was then transferred to a 96-well microtiter plate for optical density (OD) measurements at 550 nm and a reference wavelength of 650 nm. An absorbance above 0.05 indicates mitochondrial activity and cell viability.
Histology and Immunohistochemistry
Samples were fixed in 4% formaldehyde and processed for conventional paraffin embedment. Paraffin sections of 5 μm thickness were cut, deparaffinized, and rehydrated in preparation for morphological analysis (hematoxylin and eosin staining) or immunohistochemical analysis of Ki67 (MIB-1, Dako, Glostrup, Denmark), keratin 6 (Ks6.KA12, IgG1 Progen, Heidelberg, Germany), keratin 10 (DE K10, IgG1, ICN Biomedicals, Aurora, USA), and cytokeratin (LP34, IgG1 Dako, Glostrup, Denmark). For detection of Ki67, keratin 6, keratin 10, and cytokeratin a standard method for antigen retrieval on paraffin sections was performed. Sections were immersed in 0.01 M sodium citrate buffer (pH 6.0) for 30 min at 100°C followed by slowly cooling to room temperature and washing in PBS. For keratin 10 and cytokeratin only, an additional protease digestion step with 4 mg/ml pepsin in 0.2 M HCl was performed for 15 min at room temperature. Immunohistochemical analysis of melanoma-associated antigen (NKI/beteb, IgG2b, Monosan, Uden, The Netherlands) was performed using 5-μm frozen sections that were air-dried and fixed in acetone for 10 min prior to incubation with the primary antibody. After fixation and antigen retrieval, sections were washed in PBS, incubated with primary antibody for 1 h at room temperature followed by incubation with avidin-biotin-peroxidase complex (Envision+ System, Dako). All sections were washed in PBS and were counterstained with hematoxylin.
Statistical Analysis
All data are presented as mean ± SEM. The number of experiments that represent different donors with internal duplicates is stated in the figure legends. Statistical calculations were performed as follows: two-way ANOVA with Bonferroni posttest was performed using GraphPad Prism version 4.00 for Windows (GraphPad Software, San Diego, CA, USA, www.graphpad.com). Differences were significant when *p < 0.05, **p < 0.01, and ***p < 0.005 relative to control.
Results
Epidermal Cell Amplification
A coculture of keratinocytes and melanocytes was expanded from 1 cm2 of healthy skin derived from six different donors (Table 1). An average of 2.9 × 106 (±0.4 × 106) epidermal cells was isolated from each 1 cm2 of skin. After 8–9 days, cultures had reached 80–90% confluency and had amplified by a factor 1.6 ± 0.3. These subconfluent cultures were then passaged. After a total culture period of 10–14 days, the amplification factor was 5.4 ± 0.7 compared to the original cell number obtained from a 1 cm2 skin biopsy. These passage 2 keratinocytes were seeded at a density of 50,000 keratinocytes/cm2 onto Matriderm, resulting in 319 ± 79-fold increase in surface area of the original 1-cm2 skin biopsy. EC-Matriderm had a standard surface area of 150 cm2 (10 × 15 cm), which could be easily cut if required to accommodate the required seeding cell density.
Amplification of Epidermal Cells Isolated From 1 cm2 of Skin

P0: number of cells isolated from 1 cm2 of skin. P1: number of cells derived from original 1-cm2 skin biopsy after first amplification step at time first passage (day 8–9). P2: number of cells derived from original 1-cm2 skin biopsy after second amplification step at time second passage (day 10–14). Area covered: total area (cm2) that can be covered from cells amplified from the original 1-cm2 skin biopsy at time of second passage (cm2/cm2 donor skin).
Viability and Distribution of Epidermal Cells in EC-Matriderm
When testing a new carrier system it is important to determine whether the cells remain viable in the carrier and whether the cells are distributed evenly throughout the carrier. Therefore, epidermal cells were cultured in Matriderm for 3–5 days. Hereafter, multiple 3-mm biopsies were taken from different regions of EC-Matriderm and metabolic activity/cell viability was determined with the aid of an MTT assay. In total six independent donor experiments were performed in which 3–12 punch biopsies were analyzed from each 150-cm2 area of EC-Matriderm. All biopsies contained viable cells. The interdonor and intradonor variability was very low (mean OD 0.21 ± 0.01 and 0.23 ± 0.03, respectively), indicating that the protocol was highly reproducible and robust (Fig. 2).

Metabolic activity of EC-Matriderm. Metabolic activity representing cell viability of 3-mm-diameter punch biopsies derived from EC-Matriderm. Each point represents the mitochondrial activity of one biopsy. The line indicates the average value for a particular donor. The bar indicates the mean of all six donors ± SEM. No significant difference was observed between the donors.
In order to determine the distribution of epidermal keratinocytes within the EC-Matriderm, 3-mm biopsies were taken for immunohistochemical staining with anticytokeratin. Individual keratinocytes were distributed evenly over the seeding surface of the carrier (Fig. 3b). Penetration into the carrier was limited and no keratinocytes were observed on the lower surface of the carrier. Notably, no differentiating keratinocyte clones were observed, indicating that the Matriderm carrier is enriched with proliferating, undifferentiating keratinocytes.

Culture and transfer of epidermal cells onto human dermis. (a) A representative epidermal cell culture undergoing amplification. (b) Immunohistochemical staining of EC-Matriderm with anti-human cytokeratin shows distribution of keratinocytes within the Matriderm carrier. (c) Transfer of epidermal cells from EC-Matriderm (1 cm2) by placing EC-Matriderm onto acellular human dermis for 3 days. (d) The regenerating, as yet unpigmented, epidermis is visible as a yellow layer on the dermis after 2 weeks culture.
Transport Conditions for EC-Matriderm
When considering a future clinical application for EC-Matriderm it is important to predefine the transport conditions. Therefore, we determined whether EC-Matriderm could remain outside of the culture incubator for a period of 24 h without any long-term adverse effects occurring with regards to EC metabolic activity and viability (Fig. 4). When EC-Matriderm was placed under transport conditions (HBSS, 20°C, atmospheric oxygen and humidity) for 24 h, a slight decrease in metabolic activity was observed compared to parallel control cultures. However when EC-Matriderm was transferred from transport conditions back to optimum culture conditions again a full recovery was observed, indicating no long-lasting negative effects due to the transport conditions. Indeed, the metabolic activity was greater than at the beginning of the experiment, indicating that the cultures were actively growing again after transport.
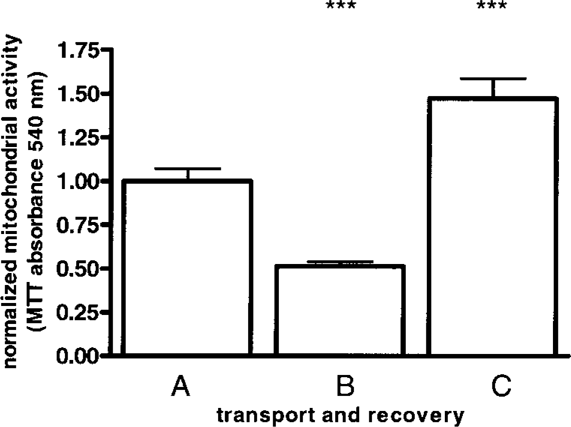
Transport conditions for EC-Matriderm. The bars represent the metabolic activity of 3-mm-diameter punch biopsies derived from EC-Matriderm obtained at different time intervals during transport and recovery. A: metabolic activity of EC-Matriderm cultured at optimal conditions before transport; B: metabolic activity of EC-Matriderm placed at ambient conditions for 24 h; and C: metabolic activity of EC-Matriderm placed at ambient conditions for 24 h and then cultured under optimal conditions for a further 48 h. The bars indicate the mean of 10 biopsies each derived from three donors ± SEM. ***p < 0.005 relative to control (A).
Transfer of Epidermal Cells From EC-Matriderm to Dermis Results in Epidermal Regeneration
Next the cell-seeded surface of EC-Matriderm was placed in contact with human dermis and cultured for 3 days air exposed (Fig. 3c). Hereafter, the EC-Matriderm was removed and it was determined whether epidermal cell transfer had occurred to such a degree that the epidermis could be regenerated. Indeed, epidermis regenerated with a 100% success rate with no epidermal blistering: no separation of the epidermis from the basement membrane (three independent donors each with internal duplicates) (Fig. 3d). Histological analysis showed that the reconstructed epidermis was fully differentiated and contained a compact basal layer of cells, a stratum spinosum, stratum granulosum, and stratum corneum (Fig. 5). Melanocytes (NKI-beteb staining) and proliferating cells (Ki67 staining) were located throughout the basal layer at a similar frequency to that found in native skin. Epidermal differentiation was normal as shown by keratin 10 immunostaining. Only keratin 6 immunostaining was clearly different in that it was expressed suprabasally in the regenerated epidermis whereas it was absent in native epidermis.

Cultured regenerated epidermis (cultured for 14 days) closely resembles healthy native skin. A fully differentiated epidermis is shown by hematoxylin and eosin staining (H/E). Immunohistochemical staining for melanocytes (anti-NKI-beteb), proliferating cells (anti-Ki67), differentiating cells (anti-keratin 6), and activated epidermis (anti-keratin 10) is shown. Scale bars: 50 μm.
Discussion
This study describes a potential novel cell culture, transport, and transfer protocol that may be suitable for delivering cultured proliferating EC to large open skin wounds. In this in vitro study, we have taken into account the amplification factor of the cultured EC, the composition of the culture medium with respect to regulatory issues, the type of transport carrier used, and the success rate of epidermal regeneration after transfer to human dermis.
This study included a slight refinement of the culture medium [compared to our previous study (7)] in order for the EC-Matriderm to comply with the regulations set by the Dutch Central Committee on Research Involving Human Subjects (CCMO) as suitable for human clinical studies, our future aim. Therefore, we first confirmed that the amplification factor of the cultured EC was still in the same order of magnitude as our previous study. Indeed, enough EC could be cultured from 1 cm2 of skin to produce 319 ± 79 cm2 EC-Matriderm, which correlated closely to our previously reported 400-fold amplification factor. This amplification factor is somewhat less than that reported by the Rheinwald and Green method (~600-fold). This is most likely to be due to the absence of 3T3 feeder cells in our method. However, it should also be noted that it takes 3–4 weeks of culture to obtain this amplification factor and a confluent sheet suitable for transplantation compared to the 2-week culture protocol described in this study (12). Our shortened culture protocol is extremely beneficial when considering treating burns patients with autologous EC.
In our study we chose Matriderm as a transport carrier for the EC. This 3D matrix is registered as a medical device and has previously been used in clinical studies for stimulating dermal regeneration in severe burns (15,26). Here we describe a potentially novel application for this commercially available artificial matrix. Seeding EC onto Matriderm makes it possible to transport and transfer keratinocytes as single proliferating, undifferentiated cells with good regenerative potential. This is favorable above transporting and transferring differentiated keratinocytes, which have low regenerative potential. Our method was reproducible and showed little inter- and intraexperimental variation with regards to location and number of viable cells within the Matriderm. Other investigators have also realized the potential of transporting single proliferating keratinocytes to the wound rather than confluent differentiated EC cultures.
A number of strategies have been used for delivering subconfluent keratinocytes onto a wound bed. One of these is to spray a suspension of epidermal cells onto the wound with the aid of a nozzle cell spray apparatus (19). Although this is a very attractive technique, it does have a number of potential disadvantages above our method. The primarily disadvantage is that keratinocytes in suspension undergo terminal differentiation if lacking cell–cell and cell–substratum contacts for even a limited period of time (<1 h) (1,9,23,28). This means that the keratinocytes cannot be transported from a culture facility outside of the hospital and that in general this technique is applied in a one-step protocol, without EC amplification. The skin is excised and epidermal cells are isolated and then sprayed directly onto the wound, all within the time allotted to surgery. Obviously this demands time and expertise unrelated to patient care from clinicians. A further drawback with spraying keratinocytes in suspension onto the wound bed is the risk of an uneven delivery, particularly if the wound bed is not entirely horizontal. The use of EC-Matriderm potentially overcomes both of these problems because the single proliferating keratinocytes do not undergo rapid terminal differentiation due to their cell–substratum contacts, and furthermore the EC-Matriderm is designed to be applied as a living dressing onto the wound bed.
Other investigators have treated burns with cultured autologous keratinocytes suspended in fibrin glue (17). Again a drawback with this method lies with the possibility of uneven delivery. A limited number of transport carrier systems have been described (6). Polyurethane wound dressings and the dermal regeneration template Integra™ have been used to transfer keratinocytes to athymic mice (29) and pigs (16,25). The eventual implementation of these carrier systems, together with our proposed carrier, will depend on future studies describing the clinical outcome of multicenter trials and the costs of the transport carrier.
An alternative method to transplant keratinocytes onto a wound bed is to construct in vitro a skin substitute and to transplant this entire construct containing living cells (keratinocytes and fibroblasts) and the dermal matrix onto the wound bed. In contrast to transport carrier systems, the dermal matrix is incorporated into the wound bed rather than being discarded after autologous cell transfer. A number of clinical studies have been described in which autologous skin substitutes have been used to treat deep burns (4,18) and chronic ulcers (10,27). Matriderm has been used as a dermal matrix in a preclinical study for the in vitro construction of a skin substitute (11). Whereas these studies look promising, the logistics around amplifying both autologous cell types (keratinocytes and fibroblasts) in a limited time period in order to construct enough skin substitutes to cover large wounds, as well as the transport conditions still remain important issues.
In this study we were able to show that EC within the EC-Matriderm were transferred to human dermis within 3 days to such an extent that fully differentiated human epidermis regenerated within 2 weeks. This 3-day transfer period was decided after considering the logistics of a future clinical application and would correspond with the time between applying the EC-Matriderm to a patient and the first dressing change after transplantation. The regenerated epidermis showed no signs of blistering (no separation of the epidermis from the basement membrane) and contained a compact basal layer of cells, stratum spinosum, stratum granulosum, and stratum corneum. Furthermore, melanocytes repopulated the basal layer indicating that EC-Matriderm has the potential to regenerate a pigmented epidermis. While keratinocyte proliferation and differentiation was normal and comparable to healthy intact skin, an increase in the activation marker keratin 6 was observed. This is not considered to be detrimental for EC-Matriderm but rather beneficial because keratin 6 is upregulated in normal wound healing and is thought to stimulate reepithelialization by promoting keratinocyte migration (21,24). Furthermore, we were able to transport EC-Matriderm for at least 24 h at ambient conditions with no long-lasting effects on metabolic activity and cell survival.
In summary, we show that EC-Matriderm contains viable, metabolically active EC cultured in a manner that permits easy transportation and that contains EC with the potential to form a pigmented reconstructed epidermis. Our in vitro study has produced a robust protocol that is currently being tested in a clinical study.
Footnotes
Acknowledgments
This research was financed in part by the Dutch Program for Tissue Engineering (STW/NWO), part by VU University Medical Centre, and part by grant No. 07.116 from the Dutch Burns Foundation. Dr. S. Gibbs and Prof. R. J. Scheper are shareholders in the Tissue Engineering Company A-SkinBV. The authors would like to thank and acknowledge Dr. B. M. E. von Blomberg and Dr. T. Rustemeyer for discussions.