
Abstract
The third-generation NOD/LtSz-scid/IL2Rγ null (NOD/SCID IL2Rγ null ) mouse represents a significantly improved xenograft model allowing high levels of human leukocyte engraftment over extended follow up. One remaining limitation of this mouse model, however, is the low level of circulating human erythrocytes. We established a practical ex vivo erythroid culture system of xenograft marrow progenitors to enrich for human erythroid progeny. At various time points after transplant, erythroid cells were easily assayed after 17 days of ex vivo culture of xenograft marrow, with nearly all nucleated cells of human origin and approximately 60% human GPA or CD71 positive. We then transplanted cord blood CD34+ cells marked with a lentiviral vector encoding green fluorescent protein (GFP). Three months later, ex vivo culture of xenograft marrow progenitors showed 41.3% of the cultured erythroid cells were positive for GFP and human CD71, and 56.2% were positive for GFP and human GPA, similar to that of circulating leukocytes at the same time point. Next, G-CSF mobilized peripheral blood CD34+ cells from a sickle cell trait subject were infused in this mouse model to determine if the hemoglobin pattern could be modeled. CD34+ cells from the sickle cell trait subject engrafted equally compared to CD34+ cells from normal subjects, establishing the sickle cell trait phenotype. Lastly, a comparison of adult-derived peripheral blood CD34+ cells and cord blood-derived CD34+ cells xenografted mice was made, and long term follow-up demonstrated a recapitulation of the fetal to adult hemoglobin switch. This approach should prove a useful tool for testing strategies for genetic manipulation of erythroid progeny and the study of hemoglobin switching.
Keywords
Introduction
The xenograft mouse model is an attractive tool to query the long-term repopulating potential of steady-state or modified human hematopoietic stem cells (HSC). The recently developed NOD/SCID IL2Rγ null mouse model has several advantages including a longer life span, a much reduced risk of developing malignancy, and very low numbers of murine T, B, NK, and mast cells. As a result, higher human cell engraftment can be accomplished than with previous NOD/SCID mouse models (9,26,31). Our group has established an alternative to irradiation conditioning regimen using parenteral busulfan that achieves high-level human cell engraftment at low mortality compared to conventional conditioning with total body irradiation (27). While sufficient numbers of human leukocyte can be achieved, circulating human erythrocytes in the xenograft peripheral blood remain low.
Ishikawa et al. previously showed that in xenografted newborn NOD/SCID IL2Rγ null mice, human red cells were detectable in the peripheral circulation at very low levels, and human erythroid progenitors were present at 9.5% in the bone marrow (BM) 3 months posttransplant (11). In our model using 7—10-week-old mice, human glycophorin A (GPA) positive erythrocytes were only detectable in peripheral blood after intraperitoneal injection of human holo-transferrin immediately after transplantation. These human red cells were present for about 3—4 weeks with the highest level at 0.1% (7). There were no human red cells circulating long term after transplantation, after human red cell infusion, or after splenectomy. The lack of human red cell output may result from one or more of several possibilities: differences in the erythroid stress response (24), low globin gene and protein expression (15,28), lack of human-specific cytokines (19), species differences between human and murine transferrin, or other anti-human inhibitory signals.
In this report, we have built upon our prior experience with human cell output in this NOD/SCID IL2Rγ null mouse model using ex vivo culture of xenograft marrow for human erythroid differentiation. This approach using common transplantation and cell culture techniques has allowed us to overcome the limitation of human red cell reconstitution in this model.
Materials and Methods
Mice
Male NOD. Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NOD/LtSz-scid/IL2Rγ null , NOD/SCID IL2Rγ null ) mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and have been reported elsewhere (10,26). Mice were 7—10 weeks old at the time of transplant. Mice were cared for according to a protocol approved by the Animal Care and Use Committee of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at the National Institutes of Health (NIH).
Human Hematopoietic Cell Preparation
Cord blood (CB) cells were obtained from healthy volunteers postpartum after written informed consent using a protocol approved by the NIDDK Institutional Review Board (IRB). Mononuclear cells (MNC) were obtained after density-gradient centrifugation using Ficoll-Paque Plus (Amersham Biotech, Piscataway, NJ) and red blood cells (RBCs) were removed using ammonium chloride potassium (ACK) lysis buffer (Quality Biological Inc, Gaithersburg, MD). CD34 enrichment was performed using CD34 biotinylated antibody and streptavidin microbeads (Miltenyi Biotec, Auburn, CA). Cells were passed through the positive selection column twice, and their purity assessed by flow cytometry using phycoerythrin (PE)-conjugated anti-human CD34 antibody (clone 563) on a FACS Calibur (all BD Biosciences, San Jose, CA).
Peripheral blood CD34+ cells (PB CD34+) were obtained from healthy adult volunteers using a protocol approved by the NIDDK IRB (12). Briefly, volunteers received 5 days of 10 μg/kg recombinant human (rHu) granulocyte colony-stimulating factor (G-CSF, Amgen, Thousand Oaks, CA). On day 5 of mobilization, PB CD34+ cells were collected by one 12—15-L leukapheresis, and CD34-positive selection was performed using the Isolex 300i automated immuno-magnetic system (Nexell, Miltenyi, Auburn, CA).
NOD/SCID IL2Rγnull Mice Transplantation
Busulfan (Busulfex, Otsuka Pharmaceutical, Rockville, MD) was diluted with phosphate-buffered saline (PBS, Biofluids, Rockville, MD) to deliver 35 mg/kg in a final volume of 200—500 μl and was injected into recipient mice intraperitoneally at least 24 h prior to the cell infusion. CB CD34+ cells (2 × 106) or PB CD34+ cells (2 × 106) were infused intravenously via the tail vein as previously described (8).
Lentiviral Vector Preparation and Transduction
Self-inactivating (SIN) human immunodeficiency virus 1 (HIV1) vector was prepared as previously described (6). We used a four-plasmid system with pCAG KGP1.1R (gag/pol), pCAG4-RTR2 (rev/tat), pCAGGSVSVG (VSV-G envelope), and pCL20c MpGFP. This vector expresses GFP under the control of MSCV-LTR-U3 promoter. Human CB CD34+ cells were prestimulated in X-VIVO 10 (BioWhittaker, Walkersville, MD) containing SCF, FLT3L, and TPO (all at 100 ng/ml, R&D Systems, Minneapolis, MN) in CH-296 (Retro-Nectin, Takara-shuzo, Japan)-coated plates for 24 h and then transduced with these vectors at a multiplicity of infection (MOI) of 50. Four days later, the GFP expression of transduced CB cells was evaluated by FACS Calibur.
Flow Cytometric Analysis for Donor Chimerism and Leukocyte Subsets
BM was harvested as previously described (8,22) and suspended in DMEM with 0.1% bovine serum albumin (BSA, Roche, Basel, Switzerland). BM and PB cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-human CD45 and PE-conjugated anti-mouse CD45; human leukocyte subsets were also stained with one of the following PE-conjugated antibodies: CD3, CD14, CD16, CD20, CD41, and CD56. Red blood cells were stained with anti-mouse TER119-FITC and anti-human glycophorin A (GPA)-PE (CD235a); erythrocyte subsets were stained with human CD45-FITC and CD71-PE. All antibodies were purchased from BD Biosciences. For the evaluation of GFP-marked human BM cell engraftment for the mice, we stained with anti-human CD45-PE.
Ex Vivo Culture of Progenitors for Human Erythroid Differentiation
To obtain erythroid output in vitro, we modified previously described erythroid culture methods (3,14,18,20,29). In the first 3 days (Fig. 1), mouse bone marrow (BM, 2 × 107 cells), human CB CD34+ (1—2 × 106 cells), or PB CD34+ cells (1—2 × 106 cells) were cultured in six-well plates in 5 ml of DMEM containing 10% fetal bovine serum (FCS) (Hyclone®, Thermo Scientific, UT), 2 mM glutamine with penicillin/streptomycin (Invitrogen, Carlsbad, CA), 100 ng/ml of rHu stem cell factor (SCF), and 10 ng/ml of rHu interleukin-3 (IL-3) (both R&D systems) at 37°C in 5% CO2. This cytokine combination preferentially differentiated human cells. In the next 7 days, nonadherent cells were collected and resuspended in 20 ml of media specific for erythroid differentiation from a modified protocol containing Dulbecco's modified essential media (DMEM, Mediatech, Herdon, VA), 30% FCS, 2% BSA, 10−5 M β-mercapto-ethanol, 10−6 M dexamethasone, 0.6 mg/ml human holo-transferrin (Sigma-Aldrich, St. Louis, MO), 2 mM glutamine with penicillin/streptomycin (Invitrogen), 100 ng/ml of SCF, 1.25 ng/ml of transforming growth factor-β (TGF-β, R&D Systems) in the medium, and 5 U/ml rHu erythropoietin (EPO, Amgen), in a 75-cm2 flask at 37°C in 5% CO2. In the final 7 days, the nonadherent cells were again collected and resuspended in the same erythroid-specific media but without TGF-β.
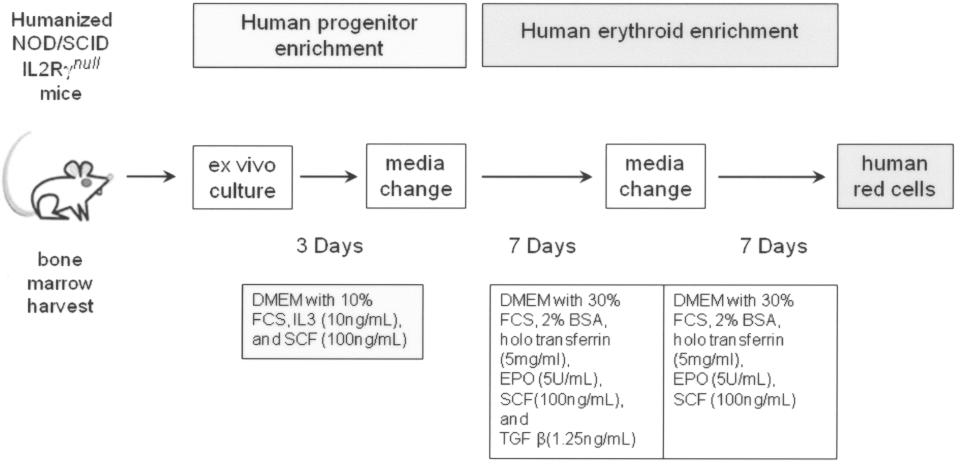
Experimental schema of ex vivo culture of progenitors for human erythroid differentiation. NOD/SCID, nonobese diabetic/severe combined immune deficiency; DMEM, Dulbecco's modified essential media; FCS, fetal calf serum; BSA, bovine serum albumin; EPO, erythropoietin; IL, interleukin; SCF, stem cell factor; TGF, transforming growth factor.
Hemoglobin Determination by Hemoglobin Electrophoresis
Hemoglobin electrophoresis was performed according to the manufacturer's procedures (Helena Laboratories, Beaumont, TX) (4). To determine the hemoglobin content from red cells, RBC pellets were lysed with water, frozen at −20°C for 5 min, and thawed at 37°C for 5 min. Then 15 μl of the supernatant was mixed with 10 μl of Hemolysate Reagent (Helena Laboratories) and run on Titan 3 cellulose acetate membranes (Helena Laboratories) at 270 volts for 40 min. After electrophoresis, the membrane was stained in Ponceau S (Helena Laboratories) for 5 min and washed with 7.5% acetic acid (Sigma) five times and ethanol five times.
Mass Spectrometry Analysis of Hemoglobin Subtypes
The products from erythroid culture were analyzed for protein content by mass spectrometry. Human GPA-positive cells were pelleted, washed twice, and lysed with water, frozen at −20°C for 5 min, and thawed at 37°C for 5 min. Proteins were analyzed using tandem HPLC-mass spectrometry (MS). Mass from eluted peptides was queried in the Mascot database, and searched for human hemoglobin variants.
Statistics
All measures of variance are presented as standard errors of the mean (SEM). Statistical analyses were performed using Student's t-test with a value of p < 0.05 deemed significant.
Results
Human Erythroid Cells Can Be Cultured From the Marrow of Humanized Mice
When murine peripheral blood was directly assayed periodically after transplantation, there was <1% human CD71+ or human GPA+ cells up to 3 months posttransplant (data not shown). We then decided to harvest BM cells from an NOD/SCID IL2Rγ null mouse 4 months after transplant with human PB CD34+ cells, and culture them in human erythroid-specific media. In the beginning of the ex vivo culture, the BM cell pellet after red cell lysis appeared white or gray, became visibly hemoglobinized at day 10, and progressed to a more red color by day 18 (Fig. 2A). When we analyzed cells for human CD71 and GPA from five other mice 1—5 months after transplantation of PB CD34+ cells, the percentage of human erythroid cells (CD71 or GPA) in culture began to increase after day 7 and plateaued between day 14 and 18 (Fig. 2B and data not shown). Without ex vivo culture, no human GPA+ cells could be detected in the circulating PB (data not shown) or BM. Ex vivo culture of murine bone marrow after transplantation of human cord blood CD34+ cells also shows similar results (Fig. 2C). To confirm that the GPA+ cells were of human origin, GPA+ RBCs from day 18 were sorted, and the hemoglobin extracts were analyzed by tandem HPLC-MS. MS revealed the presence of human α-, β-, and γ-globin chains (data not shown).

Confirmation of human-specific erythroid expansion. (A) Appearance of cell pellet before and after ex vivo culture. (B) Percent human red cells throughout ex vivo culture. The y-axis denotes the percentage of human CD71+ or GPA+ cells by flow cytometry at various time points. BM cells were collected from mice (n = 5) transplanted with 1 × 106 human PB CD34+ cells 1—6 months ago. The error bars represent SD. (C) Percent human CD45+ cells throughout ex vivo culture. The y-axis denotes the percentage of the human CD45+ cells by flow cytometry at various time points. Bone marrow cells were collected from mice transplanted with 2 × 106 human peripheral blood mobilized (PB) CD34+ cells (n = 6) or 2 × 106 cord blood (CB) CD34+ cells (n = 5) 3 months ago. The error bars represent SD.
GFP Marking Is Present Among Erythroid Progeny After Ex Vivo Culture of Humanized Murine BM
To test if this transplantation model could be employed to model gene therapy applications, human CB CD34+ cells were transduced by a lentiviral vector encoding GFP at a MOI of 50. The in vitro transduction efficiency rate was 51.7%. Transduced human CB CD34+ cells (1 × 106) were infused in each NOD/SCID IL2Rγ null mouse after conditioning with 35 mg/kg of busulfan (n = 6). The human CD45+ percentage in the PB of the humanized mice was 3.4 ± 1.7% 3 weeks posttransplant and 33.3 ± 14.3% 3 months posttransplant. Interestingly, the percentage of GFP among human CD45+ cells was 42.4 ± 5.8% 3 weeks posttransplant and increased to 79.4 ± 1.85% 3 months posttransplant. CD3+, CD14+, CD16+, CD20+, CD41+, or CD56+/GFP+ cells could also be detected in the recipient mice (data not shown).
When murine peripheral blood was directly assayed, there were no human CD71+ or human GPA+ cells 3 months posttransplant (data not shown). However, when BM was collected 3 months posttransplant in one representative mouse, 70.2% were positive for both GFP and human CD45 (Fig. 3A). After 18 days of ex vivo culture, GFP and human CD45+ cells was enriched to 79.2% (Fig. 3B—D); GFP and human CD71+, 41.3% (Fig. 3E); and GFP and human GPA+, 56.3% (Fig. 3F). We then sorted human CD71+ or GPA+ cells by using the Miltenyi magnetic selection system and confirmed an almost 100% enrichment by flow (data not shown). These experiments indicated that the genetically modified human CD34+ cells could engraft in the murine BM, and maintain the capability to differentiate to human erythroid cells ex vivo with human-specific cytokines.

Transplantation of GFP marked progenitors in NOD/SCID IL2Rγ null mice. Three months after transplantation, bone marrow (BM) from one representative mouse was harvested, and percent of green fluorescent protein (GFP) and human CD45+ cells are shown before culture (A) and after 18 days of ex vivo culture (B). GFP+ cells were visualized at day 18 under fluorescent microscope at low magnification (10x), under optical light (C) and UV light (D). Double positive cells for CD71 and GFP (E) and glycophorin-A (GPA) and GFP (F) are shown.
Recapitulation of Sickle Cell Trait in NOD/SCID IL2Rγnull Mice
We next transplanted PB CD34+ cells from healthy adults with sickle cell trait (1 × 106 cells per mouse, n = 12), or without sickle cell trait (controls, 1 × 106 cells per mouse, n = 5), in NOD/SCID IL2Rγ null mice after 35 mg/kg of busulfan. The percentage of human CD45+ cells 3 months posttransplant was similar in the PB of sickle cell trait recipients and control recipients: 13.1 ± 8.4% versus 13.9 ± 10.0%, respectively. At 1 and 3 months posttransplant, BM cells were harvested from one representative mouse from each group and cultured ex vivo. After 18 days of human erythroid differentiation, the hemoglobin composition was assessed by electrophoresis (Fig. 4). Adult hemoglobin (HbA) and sickle hemoglobin (HbS) were detected both 1 and 3 months after transplant, and the pattern was similar to that derived from sickle trait product cultured pretransplant. Interestingly, a significant amount of fetal hemoglobin (HbF) was also present at both time points, likely a reflection of the culture conditions.

Cellulose acetate hemoglobin electrophoresis gel. Lane 1 contained adult hemoglobin (HbA); lane 2, cord blood with majority of fetal hemoglobin (HbF); and lane 3, peripheral blood of sickle cell trait (HbS + HbA). Lanes 4 and 5 showed hemoglobin proteins present in the RBCs after 18 days of ex vivo culture humanized NOD/SCID IL2Rγ null mice BM. The last lane was a sample derived from PB CD34+ cells of a sickle cell trait individual after 18 days of ex vivo culture, before xenograft transplantation.
Recapitulation of Hemoglobin Switching
To determine if erythroid development occurs in a manner similar to that in humans, we performed additional experiments to examine the hemoglobin content with respect to infused cell type. First, we cultured a small aliquot of PB CD34+ or CB CD34+ cells ex vivo and determined the hemoglobin content after 18 days.
As expected, PB CD34+ cells produced more HbA, whereas CB CD34+ cells produced more HbF. The addition of TGF-β did not increase the amount HbF from PB CD34+ or CB CD34+ culture (data not shown). We then transplanted PB CD34+ and CB CD34+ cells to two groups of mice after 35 mg/kg of busulfan conditioning, harvested BM cells at various time points, and cultured BM cells ex vivo. The hemoglobin electrophoresis of the ex vivo cultured red cells showed that transplanted PB CD34+ cells consistently produced more HbA than HbF, and this proportion of HbA to HbF ratio did not change from 3 weeks to 6 months posttransplant (Fig. 5A). In contrast, transplanted CB CD34+ cells produced mostly HbF at 3 weeks, but the HbA to HbF ratio gradually reversed, similar to the ratio from PB CD34+ cells, by 6—9 months (Fig. 5B). The progressive loss of HbF synthesis over 9 months closely mirrors that which is observed in the normal hemoglobin switching, which occurs in newborns from 1 to 6 months postnatally.

Hemoglobin content from xenograft mice mimics postnatal hemoglobin switching. Bone marrow cells were collected and cultured ex vivo from xenograft mice at various time points after transplantation of peripheral blood mobilized (PB) CD34+ cells (A) or cord blood (CB) CD34+ cells (B). Cellulose acetate hemoglobin electrophoresis showed the same HbF amount at all time points in PB CD34+ group, but gradually decreasing HbF level in the CB CD34+ group.
Discussion
Humanized mice are a useful tool to assay long-term hematopoietic progenitors, and could reduce the need for the more cumbersome and expensive large-animal models. We and others have achieved high level in vivo human cell engraftment in this third-generation NOD/ SCID mouse model, which is sustained beyond 4—6 months and in multiple leukocyte subsets (granulocytes, B and T lymphocytes, and NK cells). However, human erythroid cells in murine peripheral blood remained low in vivo, despite injection with human cytokines (7). In this report, we extend our prior observations and demonstrate that human erythroid progenitors reside long term in the humanized murine bone marrow and can differentiate to sufficient numbers of mature red cells ex vivo in the presence of human-specific cytokines to allow evaluation of hemoglobin at the protein level. This straightforward approach of transplantation, marrow progenitor harvest, and ex vivo culture overcomes one limitation of the NOD/SCID IL2Rγ null xenograft model and allows for assaying erythroid progeny of human HSCs after extended follow up.
Our ex vivo culture system employs common murine transplantation techniques, reagents, and human-specific cytokines. The first 3 days of rHu IL-3 and SCF preferentially enriched human cells. The next 7 days of SCF, EPO, and TGF-β directed the differentiation of erythroid cells: SCF increased erythroblast growth (17,21,29), EPO provided the necessary erythroid cytokine, and TGF-β accelerated this entire process (1,2,32). We have previously shown that human holo-transferrin provides the iron necessary for erythroid production (7), and have incorporated this in the culture cocktail. The last 7 days of SCF and EPO served to further expand the number of mature red cells, which were positive for human CD71 or GPA. Culturing beyond the 18 days only increased the red cell number modestly; the maximal number plateaued between 3 and 4 weeks of culture (data not shown).
We then tested whether this approach could be combined with genetic modification of progenitor cells to express GFP using a SIN-lentiviral vector as a model for gene transfer approaches targeting red blood cells. The transplantation of these marked cells yielded 33% of the circulating peripheral blood leukocytes of human origin 3 months later, consistent with our prior experience. When bone marrow cells were harvested and cultured ex vivo, GFP+ and human CD45+ leukocytes could be further enriched. Furthermore, GFP+ and GPA+ red cells can be cultured using the same system, yielding sufficient quantities or red blood cells for analysis of the success of the genetic modification strategy at the protein level. The logical next step would be to substitute GFP for a gene of interest, such as β- or γ-globin, and evaluate the amount of globin expressed per vector copy in the erythroid compartment.
The usefulness of this xenograft model and ex vivo human erythroid culture also allowed us to test whether a human erythroid phenotype, such as HbS in red cells, could be reproduced. Our transplantation of G-CSF mobilized PB CD34+ cells from a sickle cell trait donor demonstrated that these cells engraft equally well, and that their ability to synthesize HbS was preserved in the xenograft bone marrow. Although not a disease model, engraftment of sickle trait HSCs in transplanted NOD/SCID mice should allow testing of genetic modification strategies aimed at correction of the hemoglobinpathies such as RNA transplicing (13) or RNA interference (25). This model may prove invaluable, as collection of mobilized CD34+ cells from individuals with sickle cell disease is associated with prohibitive toxicity (5). Additionally, other hemoglobin variants or bone marrow diseases could be modeled without transgenic techniques (16,23,30).
During the process of creating sickle cell trait mice, we discovered that the ability to make HbF was dependent on hematopoietic progenitor source. While HbF was detected from culturing PB CD34+ as shown previously, CB CD34+ cells retained substantially higher HbF synthetic potential when cultured in vitro (data not shown), or when cultured from xenograft mice shortly after transplantation (Fig. 5). This disparity between CB versus PB CD34+, however, could not be overcome even by transplantation or potent cytokine cocktails previously shown to induce HbF, such as SCF or TGF-β. The longer CB CD34+ cells remained in the xenograft bone marrow, the lower amount of HbF protein was synthesized. Eventually the HbA/HbF proportion in the CB CD34+ group approached to that of the PB CD34+ group, suggesting that the fetal to adult hemoglobin switch had occurred (30). The combination of the murine marrow microenvironment, murine cytokines, and/or gene silencing was sufficiently potent to mimic the normal postnatal human hemoglobin switching process, across a major interspecies difference.
In summary, our practical method to evaluate the erythroid compartment in the humanized NOD/SCID IL2R-γ null mice model is an important step forward and should allow for future preclinical testing of genetic modification strategies aimed at human red cells. This ex vivo human erythroid culture system also allows recapitulation of hemoglobin variants without the need for creating transgenic mice. Finally, the combination of transplantation and ex vivo culture simulated the fetal to adult hemoglobin switch, and should allow future detailed investigation of this important human postnatal transition.
Footnotes
Acknowledgments
This work was supported by the Intramural Research Program of NIDDK and NHLBI at the NIH.